Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2014; 4(7):745-760. doi:10.7150/thno.7811 This issue Cite
Research Paper
Dimerization of a Phage-Display Selected Peptide for Imaging of αvβ6- Integrin: Two Approaches to the Multivalent Effect
Ajay N. Singh1, Michael J. McGuire2, Shunzi Li2, Guiyang Hao1, Amit Kumar1, Xiankai Sun1,3,4 ![]() , Kathlynn C. Brown2,4
, Kathlynn C. Brown2,4 ![]()
1. Department of Radiology,
2. Department of Internal Medicine,
3. Advanced Imaging Research Center, and
4. The Simmons Comprehensive Cancer Center, University of Texas Southwestern Medical Center, Dallas, Texas.
Received 2013-10-3; Accepted 2013-10-10; Published 2014-5-15
Citation:
Singh AN, McGuire MJ, Li S, Hao G, Kumar A, Sun X, Brown KC. Dimerization of a Phage-Display Selected Peptide for Imaging of αvβ6- Integrin: Two Approaches to the Multivalent Effect. Theranostics 2014; 4(7):745-760. doi:10.7150/thno.7811. https://www.thno.org/v04p0745.htm
Other stylesAbstract

The integrin αvβ6 is an emerging biomarker for non-small cell lung cancer (NSCLC). An αvβ6-binding peptide was previously selected from a phage-displayed peptide library. Here, we utilize a multivalent design to develop a peptidic probe for positron emission tomography (PET) imaging of αvβ6+ NSCLC tumors. Multimeric presentation of this peptide, RGDLATLRQL, on a bifunctional copper chelator was achieved using two approaches: dimerization of the peptide followed by conjugation to the chelator (H2-D10) and direct presentation of two copies of the peptide on the chelator scaffold (H2-(M10)2). Binding affinities of the divalent peptide conjugates are four-fold higher than their monovalent counterpart (H2-M10), suggestive of multivalent binding. PET imaging using the bivalent 64Cu-labeled conjugates showed rapid and persistent accumulation in αvβ6+ tumors. By contrast, no significant accumulation was observed in αvβ6- tumors. Irrespective of the dimerization approach, all divalent probes showed three-fold higher tumor uptake than the monovalent probe, indicating the role of valency in signal enhancement. However, the divalent probes have elevated uptake in non-target organs, especially the kidneys. To abrogate nonspecific uptake, the peptide's N-terminus was acetylated. The resultant bivalent probe, 64Cu- AcD10, showed drastic decrease of kidney accumulation while maintaining tumor uptake. In conclusion, we developed an αvβ6-integrin specific probe with optimized biodistribution for noninvasive PET imaging of NSCLC. Further, we have demonstrated that use of multivalent scaffolds is a plausible method to improve library selected peptides, which would be suboptimal or useless otherwise, for imaging probe development.
Keywords: Lung Cancer, Molecular Imaging, Positron Emission Tomography, Integrin, Peptide.
Introduction
Non-small cell lung cancer (NSCLC) is the leading cause of cancer related mortality worldwide, accounting for ~1.2 million deaths each year (1). The lethality of NSCLC is due in part to diagnosis at advanced stages, tumor heterogeneity, and rudimentary tumor classification (2, 3). The advent of targeted therapy has changed the paradigm of cancer management by targeting aberrant molecular and cellular changes in order to inhibit tumor growth and progression. Noninvasive diagnostic tools that reveal molecular features of a NSCLC tumor are needed to provide personalized molecular medicine for improved therapeutic response and patient survival.
Chest X-ray and Computed Tomography (CT) are the primary diagnostic tools to identify lung lesions. Inherently anatomical imaging techniques, CT and X-ray cannot provide molecular profiles of heterogenic NSCLC tumors (4). By contrast, PET is well suited for molecular profiling due to its high sensitivity and capability of absolute imaging quantification (5, 6). Indeed, PET-CT imaging, which combines the anatomic precision of CT with the functional imaging of PET, has significantly improved detection of tumors, and is increasingly used in clinical setting for noninvasive diagnosis and staging of cancer (4-6). By far, 2-deoxy-2-18F-fluorodeoxyglucose (18F-FDG) is the most widely used PET tracer in diagnosis, staging, and monitoring of NSCLC (4, 7). However, this glucose consumption-based PET tracer plays a limited role in distinguishing tumor masses based on their molecular alterations.
Design of molecular PET imaging probes relies on judicial selection of a cellular biomarker for early detection, treatment planning, and monitoring of disease progression. The αvβ6 integrin, a transmembrane protein, is a unique biomarker which is essentially absent in normal adult epithelium but has shown de novo expression during tissue remodeling, epithelial repair, and tumor growth (8, 9). Widespread up-regulation of αvβ6-integrin is found in early stage NSCLC and is predictive of reduced survival time for patients. Furthermore, αvβ6 expression increases during the epithelial-mesenchymal transition, the process in which cells lose their epithelial phenotypes to become motile, implicating a role in metastasis (10-12). The differential expression of αvβ6-integrin in epithelial based tumor cells over normal cells suggests αvβ6-integrin probes will provide a high tumor-to-normal tissue contrast. Thus, developing a αvβ6-integrin targeted PET imaging probe provides an exciting diagnostic tool to detect and classify an aggressive NSCLC phenotype. Significantly, αvβ6-integrin expression is also observed in several other epithelial based tumors, suggesting αvβ6-imaging probes might find wide utility beyond NSCLC (13-16).
Specific αvβ6-ligands have been identified from natural sources as well as from combinatorial peptide libraries (17-19). Use of these ligands as PET probes has met with mixed success. Improvements in ligand affinity, in vivo stability, and tumor specificity are desired. Recently we reported the selection of a peptide, H2009.1 (sequence: RGDLATLRQL), as an αvβ6-integrin specific ligand (20). The isolated monomeric peptide displays high binding affinity (9 nM) and selectivity towards αvβ6 integrin over other integrins (8, 20). The peptide was selected from a pIII phage displayed library in which the peptide is displayed in 3-5 copies at the tip of the bacteriophage. As such, the selected peptide most likely interacts with the target receptor in a multivalent fashion. Indeed, multimerization of H2009.1 improves its affinity for αvβ6-expressing cells by 800-fold (20).
Multivalent presentation of a ligand is a well-accepted approach to increase the biological potency of a ligand through enhanced ligand-receptor interaction, and in some cases, cellular internalization by surface receptor oligomerization (21, 22). Peptide multimerization can also improve peptide stability and extend its in vivo half-life (23, 24). In the field of molecular imaging, the multivalent effect has been exploited for imaging signal enhancement in order to achieve the desired signal-to-noise level (25, 26). Generally, a multimerized targeting vector is conjugated on a bifunctional chelator (BFC) to render the imaging probe design multivalent effect (27-30). Alternatively, we recently reported a novel and versatile BFC scaffold (BFCS) design, which provides a simple and effective way of imparting multivalency to imaging probes (25, 26). In this work, we utilized these two complimentary approaches to present two copies of H2009.1-10mer at different layers of a BFCS for imaging of integrin αvβ6. One is a peptide based multivalency constructed upon the dimerization of the H2009.1-10mer sequence (H2-D10, and H2-AcD10, Scheme 1). The other is a BFCS based multivalency achieved by anchoring the H2009.1-10mer sequence at the two peripheral carboxylate groups of the BFCS (H2-(M10)2, Scheme 1). The resulted divalent conjugates were evaluated and compared to the monovalent conjugate H2-M10 (Scheme 1). The αvβ6 imaging properties of the peptide conjugates when labeled with 64Cu were assessed using H2009 (αvβ6+) and H460 (αvβ6-) tumor models established in SCID mice.
Methods and Materials
General Methods: All reactions were carried out under N2 atmosphere in degassed, dried solvents. Commercially available starting materials were used without further purification unless otherwise stated. Aqueous solutions were prepared with Milli-Q water (18 MΩ-cm) obtained from a Millipore Gradient Milli-Q water system (Billerica, MA). Matrix-assisted laser desorption/ionization (MALDI) mass spectra were acquired on an Applied Biosystems Voyager-6115 mass spectrometer. Radiolabeled conjugates were purified by Light C-18 Sep-Pak cartridges (Waters, Milford, MA).
Scheme 1
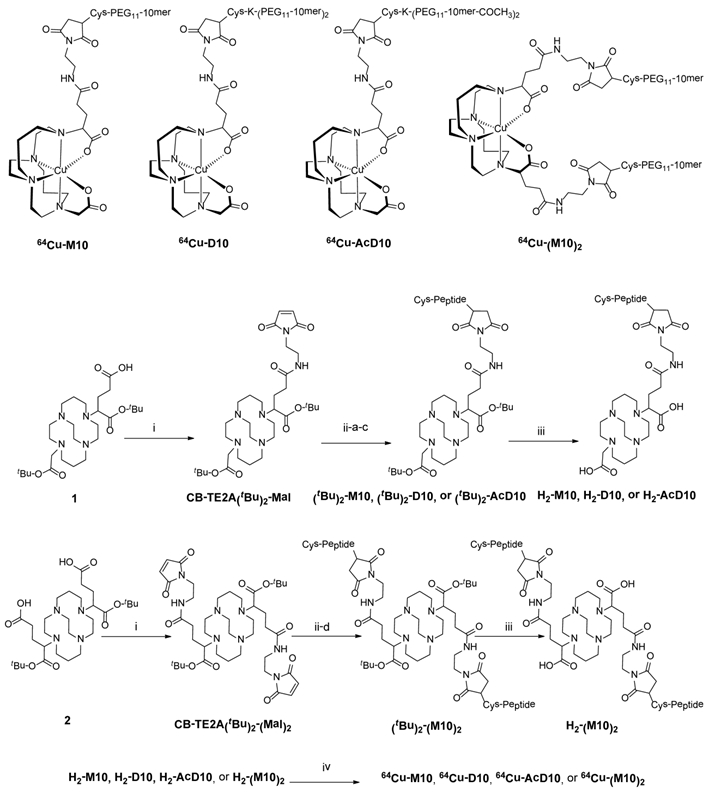
Structures of PET probes, 64Cu-M10, 64Cu-D10, 64Cu-AcD10, and 64Cu- (M10)2. Syntheses of monomer (64Cu-M10), two peptide based divalent (64Cu-D10, and 64Cu-AcD10), and a scaffold based divalent (64Cu-(M10)2) are shown. (i) N-(2-aminoethyl)maleimide, HATU, DIPEA; (ii-a) H2009.1-monomer; (ii-b) H2009.1-dimer; (ii-c) acetylated H2009.1-dimer; (ii-d) H2009.1-monomer; 10mM PBS (pH = 7.0), 10mM EDTA; (iii) TFA (95%); (iv) 64CuCl2, 0.4 M NH4OAc.
The 1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-cyclam),(31) α-bromoglutaric acid-1-tertbutylester- 4-benzyl ester, compound 1, compound 2,(25) Cys-PEG11-H2009.1-10mer), Cys-K-(PEG11- H2009.1-10mer)2, and Cys-K-(PEG11-H2009.1-10mer-COCH3)2) were synthesized by standard Fmoc solid phase peptide synthesis according to published procedures (20). All peptides were >95% as determined by analytical HPLC. Copper-64 (t1/2 = 12.7 h, β+ (17.4%; Eβ+max = 0.656 MeV); EC (43%); β- (39%)) in 0.1 N HCl was purchased from Washington University School of Medicine in St. Louis or the University of Wisconsin at Madison.
Cell Lines and Animals: H2009 and H460 cell lines were provided by the Hamon Center for Therapeutic Oncology Research (UT Southwestern Medical Center) and cultured according to established conditions (32). The cell lines were tested for Mycoplasma infection and DNA fingerprinted before use. Animal protocols were approved by the Institutional Animal Care and Use Committee at UT Southwestern which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The protocols adhere to standards set forth by the Animal Welfare Act and the US government principals regarding the care and use of laboratory animals. To establish tumor xenografts, NOD/SCID mice were injected with 5 × 106 H2009 or H460 cells in the upper flank towards the shoulder. Tumors were grown to 100 - 200 mm3 (tumor volume = ½(length × width2) before imaging (33).
HPLC Methods: Peptide conjugates were purified by HPLC using a Waters 600 Multisolvent Delivery System. The mobile phase consisted of H2O with 0.1% trifluoroacetic acid (TFA, solvent A) and acetonitrile with 0.1% TFA (solvent B). Peptide conjugate purification was performed on a semi-preparative XTerra RP18 Column (250 × 10 mm) with a gradient of 0% B to 100 % B in 50 min at a flow rate of 4.0 mL/min. Analytical HPLC was performed on an XTerra RP18 column (150 × 4.6 mm) with a gradient of 0 % B to 100 % B in 50 min at a flow rate of 1.0 mL/min.
Synthesis of H2-M10: CB-TE2A(tBu)2-Mal (1.8 mg, 2.8 μmol) and Cys-PEG11-10mer peptide (1.3 mg, 0.7 μmol) were reacted in a 10 mM PBS containing 10 mM ethylendiaminetetraacetic acid (EDTA, 500 μL, pH 7.0) at room temperature for 12 h under N2. The conjugate was purified by semi-preparative reverse-phase HPLC. Lyophilization yielded (tBu)2-M10 as white solid (1.2 mg; yield: 70%). MALDI-TOF/MS: 2492.71 [M+H]+. Calc'd MS: 2491.44 The protected conjugate, (tBu)2-M10, was dissolved in 95% of TFA and stirred at room temperature for 12 h. Solvent was evaporated and the residue was purified by semi-preparative reverse-phase HPLC. The collected fraction of multiple runs were pooled and lyophilized to give H2-M10. MALDI-TOF/MS: 2380.76 [M+H]+. Calc'd MS: 2379.23
Synthesis of H2-D10 and H2AcD10: To a mixture of CB-TE2A(tBu)2-Mal (1.0 mg, 2.1 μmol) and Cys-K-(PEG11-10mer)2 peptide (3.0 mg, 0.7 μmol) was added 10 mM PBS solution containing 10 mM EDTA (500 μL, pH 7.0). The reaction mixture was stirred at RT for 12 h under N2. The reaction mixture was purified by semi-preparative reverse-phase HPLC and lyophilized to give protected (tBu)2-D10 as white solid (1.8 mg; yield: 56%). MALDI-TOF/MS: 4967.07 [M+H]+. Calc'd MS: 4966.46. The (tBu)2-D10, was dissolved in 95% of TFA and stirred at RT for 12 h to remove the tBu protecting groups. Semi-preparative reverse-phase HPLC followed by lyophilization to yield H2-D10. MALDI-TOF/MS: 4847.23 [M+H]+. Calc'd MS: 4846.89
The acetylated conjugate was prepared following the same procedure using CB-TE2A(tBu)2-Mal (1.0 mg, 2.1 μmol) and Cys-K-(PEG11-10mer-COCH3)2) peptide (3.0 mg, 0.68 μmol). Protected (tBu)2-AcD10 was obtained as a white solid (2.5 mg; yield: 73%). MALDI-TOF/MS: 5048.15 [M+H]+. Calc'd MS: 5047.49. After deprotection, H2-AcD10 was obtained in quantitative yield. MALDI-TOF/MS: 4935.46 [M+H]+. Calc'd MS: 4934.85
Synthesis of H2(M10)2: To the mixture of CB-TE2A(tBu)2-(Mal)2 (0.2 mg, 0.23 μmol) and Cys-PEG11-10mer peptide (1.3 mg, 0.7 μmol) was added 10 mM PBS solution containing 10 mM EDTA (500 μL, pH 7.0). The reaction mixture was stirred for 12 h under N2. The reaction mixture was purified by semi-preparative reverse-phase HPLC. The collected fractions from multiple runs were pooled and lyophilized to give protected (tBu)2-(M10)2 (0.67 mg; yield: 63%). MALDI-TOF/MS: 4529.62 [M+H]+. Calc'd MS: 4528.53. The protected conjugate, (tBu)2-D10, was dissolved in 95% of TFA and stirred at room temperature for 12 h. Solvent was evaporated and the residue was purified by semi-preparative reverse-phase HPLC. Lyophilization afforded H2-(M10)2 as white solid in quantitative yield. MALDI-TOF/MS: 4414.57 [M+H]+. Calc'd MS: 4413.37
Radiolabeling of H2M10, H2D10, H2AcD10, and H2(M10)2 with 64Cu: To a 100 μL vial containing 5-10 μg of a respective conjugate in 50 μL of 0.4 M NH4OAc buffer (pH = 6.5), 1-2 mCi of 64CuCl2 was added. The reaction mixture was shaken at 75ºC for 30 min. Then, 2 μL of 5 mM EDTA was added to the reaction mixture, which was allowed to incubate for 5 min. Purification of 64Cu-labeled conjugate was carried out on a Sep-Pak C-18 light cartridge preconditioned with 10 mL EtOH followed by 10 mL H2O. After thorough rinsing (3 × 3 mL, water) of the cartridge, the 64Cu-labeled conjugate was eluted by an ethanol-PBS (10 mmol) mixture (70:30). The product was analyzed on instant thin-layer chromatography (ITLC-SG) plates (Pall Life Sciences, East Hills, NY) read by a Rita Star Radioisotope TLC Analyzer (Straubenhardt, Germany). The radiochemical purity of the 64Cu-labeled conjugates after cartridge purification was > 97% as determined by radio-HPLC. The overall radiochemical procedure including the synthesis and purification steps took less than 45 min and gave a decay-corrected radiochemical yield of > 70%. The specific activity of the purified 64Cu-labeled conjugates was in the range of 15-30 GBq/μmol.
Small Animal PET/CT Imaging: Imaging studies were performed on a Siemens Inveon Multimodality PET/CT system. One hour prior to imaging, each mouse bearing a H2009 or H460 tumor was injected with 100 - 125 µCi of a 64Cu labeled conjugate in 100 µL of saline via the tail vein (n=3 for each experiment). Ten minutes prior to imaging, the animals were anesthetized using 3% isofluorane in O2 at room temperature until vital signs stabilized. The animal was placed onto the imaging bed under 2% Isofluorane anesthesia for the duration of the imaging. At each time point (1 h, 4 h, and 24 h) post-injection (pi.), a CT scan was performed (6 min), immediately followed by a static PET scan (15 min). The CT imaging was acquired at 80kV and 500 μA with a focal spot of 58 μm. Total rotation of the gantry was 360o with 360 rotation steps obtained at an exposure time of approximately 180 ms/frame. The images were attained using CCD readout of 4096 × 3098 with a binning factor of 4 and an average frame of 1. Under low magnification the effective pixel size was 103.03 μm. The CT images were reconstructed with a down sample factor of 2 using Cobra Reconstruction Software. PET images were reconstructed using Fourier Rebinning and Ordered Subsets Expectation Maximization 3D (OSEM3D) algorithm. Reconstructed CT and PET images were fused and analyzed using Inveon Research Workplace (IRW) software. For quantification, regions of interest were placed in the areas expressing the highest 64Cu-labeled conjugate activity as determined by PET and guided by visual inspection of CT images. The resulting quantitative data were expressed in % injected dose/g (%ID/g).
Serum stability test: In vitro stability test was performed in rat serum. Briefly, 64Cu labeled conjugate (0.74 MBq, 5 μL) was added into 100 μL of rat serum and incubated at 37 °C (n = 3). After 1 h, 4 h and 24 h incubation, 50 μL of the sample was taken out and mixed with 250 μL of ethanol. The solution was vortexed and centrifuged for 5 min at 21,000 g. Both the pellet and the supernatant were counted and the supernatant was further analyzed by radio-HPLC.
Peptide Competition Assay: Binding affinity of the peptide-conjugates (H2-M10, H2-D10, H2-AcD10, and H2-(M10)2) to H2009 cells was determined as previously described by varying the peptide concentration by factors of 10 ranging from 0.1 to 1000 nM (34). Peptide stock solutions were prepared in phosphate buffered saline (pH 7.4) and concentrations were determined by 3-(4-carboxybenzoyl)quinoline-2-carboxaldehyde assay (CBCQA assay, Invitrogen). Peptide stock solutions were diluted in PBS/BSA containing 108 particles of the H2009.1 phage clone which displays the parental H2009.1 peptide. Chloroquine (0.1 mM) and 1 × protease inhibitor (Roche) were included to minimize phage degradation. The peptide-phage solution was incubated on 90% confluent H2009 cells in a 12-well tissue culture dish for 10 minutes. Cells were then washed 4 times with PBS/BSA and 2 times with 0.1 M HCl-Glycine in 0.9% saline (pH 2.2). Cells were lysed in 30 mM Tris-HCl, (pH 8.0) by freeze-thaw. Cell-associated fractions were diluted, mixed with K91 E. coli and colony forming units were determined. Peptide blocking is defined as the output phage to input phage ratio in the presence of the peptide compared to the same ratio without added peptide.
Binding Assays on Purified Integrins: The dimeric H2009.1-10-mer and scrambled H2009.1-10-mer peptides were synthesized in house with a biotinylated lysine incorporated into the dimeric core. cRGDfK-PEG-PEG-biotin and RADfK-PEG-PEG-biotin were purchased from Peptides International. Purified integrins (R & D systems) were resuspended from lyophilized powder in PBS+ at 1 µg/ml and absorbed onto Nunc MaxiSorp flat bottom polystyrene plates (96 well) overnight at 4oC.Peptide solutions at 0, 200, 100, 50, 20, 10, 5, 2, 1, 0.5, 0.2, and 0.1 nM were added to the wells containing the integrin proteins. After 30 minute incubation at room temperature, the wells were washed 3-times with PBS. Peptide capture was measured with Avidin-HRP and developed with TMB. Specific binding was determined using scramble H2009.1 dimeric peptide or cRADfY as controls. Dissociation constants were determined using nonlinear regression in GraphPad Prism using assuming a one-site binding model.
Flow Cytometry on Intact Cells: Peptide binding to cells by flow cytometry was performed as previously described (29). Cells were plated in 12 well tissue culture plates and cultured 24 to 48 hours prior to each experiment. Steptavidin-phycoerythrin (BDBiosciences, San Jose, CA) was incubated with H2009.1-dimeric peptide containing 1 biotin at 20 nM peptide concentration in PBS for 30 minutes at room temperature. Excess biotin binding sites on streptavidin were then quenched by the addition of an equal volume of RPMI. Culture medium was removed from each well and the indicated concentration of the peptide added and incubated with cells for 1 hour at 37°C. Peptide-SAPE was removed and the cells were washed 3 times in PBS, 2 times in acid wash pH 2.2, and removed from the well in PBS containing 10 mM EDTA. Peptide-mediated SAPE uptake in cells was determined by flow cytometry on a Beckman Coulter Cell Lab Quanta (Beckman Coulter Inc., Indianapolis, IN). Collected data was evaluated using WinMDI v2.9 flow analysis software. The arbitrary binding units reflect the mean fluorescence intensity.
Statistical analysis: Quantitative data were expressed as the mean ± SD. Unpaired t test (two-tailed, confidence intervals: 95%) was performed using GraphPad Prism. P values of <0.05 were considered statistically significant.
Results
Synthesis
The chelator 4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane (CB-TE2A) forms a thermodynamically and kinetically stable complex with Cu(II). In our design, the peptide attachment is through the peripheral carboxylate groups (Scheme 1). As such, the unique feature of CB-TE2A is preserved: the two inner carboxylates to form an octahedral complex with Cu(II) along with the four nitrogen atoms of the macrocycle. This results in a stable and neutral copper complex to which multiple ligands can be attached.
Synthetic routes to the monovalent (H2-M10) and divalent conjugates (H2-(M10)2, H2-D10, and H2-AcD10), (Scheme 1) consist of three parts, i) synthesis of BFC scaffolds 1 and 2,(25) ii) formation of maleimide derivatives: CB-TE2A(tBu)2-Mal and CB-TE2A(tBu)2-(Mal)2, and iii) conjugation of the H2009.1-10mer peptide to the BFCS, CB-TE2A(tBu)2-Mal or CB-TE2A(tBu)2-(Mal)2, followed by deprotection of the α-carboxylate groups to provide H2-M10, H2-(M10)2, H2-D10, and H2-AcD10 as final products.
Scaffolds 1 and 2 were synthesized in quantitative yield by reaction of CB-cyclam with α-bromoglutaric acid-1-tertbutylester-4-benzyl ester (25). They were further functionalized with one or two maleimide groups, to yield CB-TE2A(tBu)2-Mal and CB-TE2A(tBu)2-(Mal)2 in 34 and 45% yields, respectively. The maleimide moiety reacts selectively with thiols, thus minimizing issues with chemical regiospecificity upon conjugation. Also this imparts flexibility in the design of peptides as cysteine is not a common amino acid and can be conveniently incorporated into a peptide sequence as handle for further modification. In addition, this chemistry keeps the peptide sequence intact, which allows the multivalent presentation of the peptide to be conserved on the BFCS as in the original phage clone.
The maleimide intermediate, CB-TE2A(tBu)2-Mal, was conjugated with one equivalent of Cys-PEG11-10mer to provide (tBu)2-M10. Similarly, the peptide dimers (Cys-K-(PEG11-10mer)2 and Cys-K-(PEG11-10mer-COCH3)2) were conjugated to CB-TE2A(tBu)2-Mal, to afford the protected divalent conjugates (tBu)2-D10 and (tBu)2-AcD10, respectively. The BFCS based dimer, (tBu)2-(M10)2 was synthesized by conjugation of CB-TE2A(tBu)2-(Mal)2 with two equivalents of Cys-PEG11-10mer peptide. Finally, deprotection of t-butyl group afforded the monovalent and divalent peptide conjugates in quantitative yield. All four conjugates (H2-M10, H2-(M10)2, H2-D10, and H2-AcD10) were successfully labeled with 64Cu to give 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-(Ac)D10. The overall radiochemical procedure, including synthesis and purification, was completed in less than 1 h, with the specific radioactivity of 64Cu-labeled conjugates in the range of 15-30 GBq/µmol.
Binding Assays
The in vitro αvβ6 binding affinities of H2-M10, H2-(M10)2, H2-D10, and H2-AcD10 were determined by our previously described method using αvβ6 integrin-positive H2009 human NSCLC cells (20, 34). All the conjugates inhibited the binding of H2009.1 peptide phage to the αvβ6 integrin-positive H2009 cells in a dose-dependent manner (Figure 1). The IC50 values of the parental H2009.1 peptides, which represent their concentrations required to inhibit 50% of the H2009.1 peptide phage binding, were 9.2 nM (monomeric 10mer), and 0.56 nM ((10mer)-PEG11)2-K-Cys), respectively. The IC50 values of the synthesized peptide conjugates were 4.5 (H2-M10), 0.89 nM (H2-(M10)2, 0.85 nM (H2-D10), and 1.3 nM (H2-AcD10)), indicating that the conjugation of the H2009.1-10mer peptides to the CB-TE2A scaffolds has minimal effect on the peptide's binding ability. The improved affinity of H2-M10 compared to the parental monomeric 10mer may be due to the addition of the PEG linker. All three dimers showed the anticipated multivalent effect as measured by the multivalent enhancement ratio (MVE) - the fold of IC50 value decrease of the dimers as compared to H2-M10: 5.1 for H2-(M10)2, 5.3 for H2-D10, and 3.5 for H2-AcD10. Surprisingly, H2-(M10)2 and H2-D10 displayed virtually the same binding affinity. This indicates that the peptide's multivalent presentation format is not as critical as we expected in our αvβ6 imaging probe design.
Serum stability
The serum stability of 64Cu labeled conjugates was evaluated by incubating the 64Cu labeled conjugate in rat serum at 37°C. After 1, 4 and 24 h, an aliquot was removed and the serum proteins were precipitated in ethanol. The supernatants were analyzed by radio- HPLC. At 1 h post incubation, serum stability of all four conjugates was in the range of 60-73%. A significant fraction of the multivalent conjugates remains intact (64Cu-D10 (35%), 64Cu-AcD10 (33%) and 64Cu-(M10)2 (38%)) at 4 h. As expected, the multivalent conjugates (60-73% at 1h, 33-38% at 4 h) showed higher serum stability than the monovalent conjugate (55% and 20% at 1 and 4 h). It is important to note that all the divalent conjugates showed similar stability irrespective of their design. Surprisingly, acetylation of the amino terminus of the peptide did not increase the serum stability of the peptide according to this assay. However, rat serum has notorious endopeptidase activity which would not be blocked by acetylation (35).
MicroPET Imaging
The feasibility of αvβ6-integrin targeted NSCLC tumor detection in vivo was evaluated in SCID mice bearing H2009 (αvβ6+) xenografts. Effect of multivalency on imaging signal amplification was evaluated by comparing the tumor uptake of divalent probe (64Cu-(M10)2) with the monovalent one (64Cu-M10). As summarized in Table 1, 64Cu-M10 showed moderate H2009 tumor uptake (0.55 ± 0.17 %ID/g) at 1 h p.i., but the tumor became barely visible at 4 h (0.33 ± 0.11 %ID/g) and 24 h p.i. (0.28 ± 0.11%ID/g). By contrast, the scaffold based divalent probe, 64Cu-(M10)2, showed an approximate 3-fold tumor uptake increase (p < 0.01) at all three time points (1.6 ± 0.20 %ID/g, 0.85 ± 0.19 %ID/g, and 0.79 ± 0.03 %ID/g at 1 h, 4 h, and 24 h p.i., respectively) (Figure 2, Table 1), supporting the premise of signal enhancement through dimerization of the H2009.1-10mer peptide. It is noteworthy that although the peptide based (64Cu-D10) and scaffold based (64Cu-(M10)2) divalent probes are considerably different in their peptide's spatial orientations, they displayed a similar level of tumor signal enhancement at all three time points (Figure 2, Table 1). This observation is consistent with their binding affinity measurements.
Figure 1
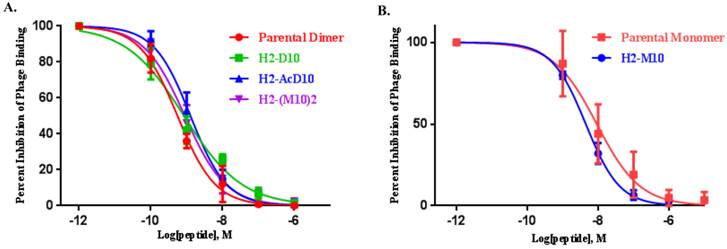
The integrin αvβ6 binding affinities of (A) H2009.1-peptide dimer, H2-(M10)2, H2-D10, and H2-AcD10, (B) H2009.1-peptide monomer and H2-M10 measured by a competitive cell-binding assay using H2009.1 peptide phage as the integrin-specific competitor. The IC50 values were calculated to be 9.2 nM (H2009.1-monomer, 95% confidence interval: LogIC50 -8.244 to -7.831, Goodness of fit (R2): 0.9932 ), 0.56 nM (H2009.1-dimer, 95% confidence interval: LogIC50 -9.336 to -9.142, Goodness of fit (R2): 0.9978), 4.5 nM (H2-M10, 95% confidence interval: LogIC50 -8.421 to -8.275, Goodness of fit (R2): 0.9993), 0.89 nM (H2-(M10)2, 95% confidence interval: LogIC50: -9.151 to -8.947, Goodness of fit (R2): 0.9983), 0.85 nM (H2-D10, 95% confidence interval: LogIC50 -9.345 to -8.800, Goodness of fit (R2): 0.9904), and 1.3 nM (H2-AcD10, 95% confidence interval: LogIC50 -9.002 to -8.783, Goodness of fit (R2): 0.9978).
Figure 2
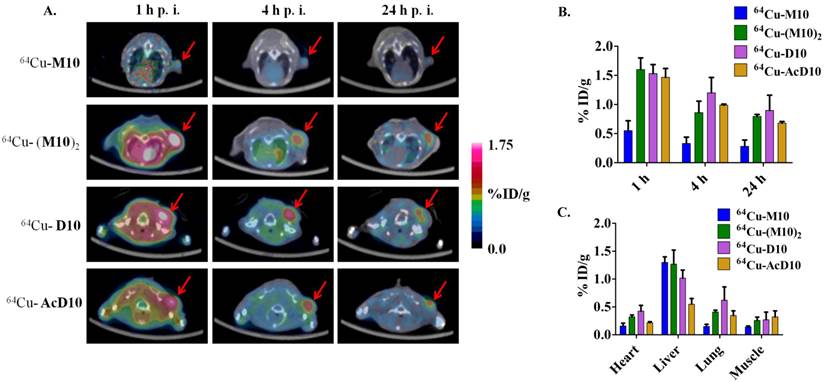
(A) Decay-corrected trans-axial PET images of 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10 in SCID mice bearing H2009 tumor at 1 h, 4 h, and 24 h p.i. Tumors are indicated by red arrow. (B) Comparative tumor uptake (%ID/g) of 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10 at 1 h, 4 h, and 24 h p.i. (C) Uptake of 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10 in other organs of interest at 24 h p.i. Data are presented as %ID/g ± s.d. (n = 3).
Table 1
Uptake 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10 at 1 h, 4 h and 24 h p.i. in major organs and tumor of H2009 tumor bearing mice determined by quantitative PET imaging analysis. Data are presented as %ID/g ± s.d. (n = 3).
| 64Cu-M10 | 64Cu-(M10)2 | ||||||
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | ||
| Tumor | 0.55 ± 0.17 | 0.33 ± 0.11 | 0.28 ± 0.11 | 1.6 ± 0.20 | 0.85 ± 0.19 | 0.79 ± 0.03 | |
| Kidney | 6.83 ± 0.35 | 6.23 ± 0.32 | 3.50 ± 0.36 | 53.6 ± 8.68 | 46.25 ± 10.39 | 27.33 ± 7.3 | |
| Heart | 1.02 ± 0.07 | 0.42 ± 0.06 | 0.16 ± 0.05 | 1.56 ± 0.15 | 0.71 ± 0.03 | 0.32 ±0.03 | |
| Liver | 2.03 ± 0.23 | 1.53 ± 0.21 | 1.30 ± 0.10 | 2.13 ± 0.73 | 1.9 ± 0.00 | 1.26 ± 0.25 | |
| Lung | 0.66 ± 0.08 | 0.33 ± 0.11 | 0.15 ± 0.04 | 1.66 ± 0.15 | 0.72 ± 0.02 | 0.40 ± 0.03 | |
| Muscle | 0.30 ± 0.02 | 0.12 ± 0.02 | 0.14 ± 0.02 | 0.87 ± 0.04 | 0.30 ± 0.02 | 0.25 ± 0.05 | |
| Tumor/Lung | 0.83 ± 0.18 | 1.0 ± 0.15 | 1.86 ± 0.11 | 1.0 ± 0.25 | 1.2 ± 0.19 | 1.9 ± 0.04 | |
| 64Cu-D10 | 64Cu-AcD10 | ||||||
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | ||
| Tumor | 1.53 ± 0.15 | 1.2 ± 0.26 | 0.89 ± 0.26 | 1.46 ± 0.15 | 0.98 ± 0.01 | 0.68 ± 0.02 | |
| Kidney | 43.46 ± 1.8 | 39.3 ± 5.66 | 21.36 ± 0.57 | 18.8 ± 2.2 | 19.06 ± 3.3 | 5.4 ± 0.87 | |
| Heart | 1.26 ± 0.37 | 0.79 ± 0.27 | 0.42 ± 0.10 | 0.78 ± 0.05 | 0.49 ± 0.4 | 0.21 ± 0.01 | |
| Liver | 2 ± 0.1 | 1.56 ± 0.11 | 1.01 ± 0.14 | 1.08 ± 0.11 | 0.72 ± 0.09 | 0.55 ± 0.10 | |
| Lung | 1.86 ± 0.20 | 0.99 ± 0.18 | 0.62 ± 0.23 | 1.33 ± 0.23 | 0.65 ± 0.03 | 0.34 ± 0.08 | |
| Muscle | 0.80 ± 0.11 | 0.40 ± 0.03 | 0.27 ± 0.13 | 0.83 ± 0.14 | 0.42 ± 0.05 | 0.32 ± 0.10 | |
| Tumor/Lung | 0.8 ± 0.25 | 1.2 ± 0.31 | 1.4 ± 0.34 | 1.1 ± 0.27 | 1.51 ± 0.03 | 2.0 ± 0.08 | |
It must be pointed out that the higher tumor uptake of 64Cu-(M10)2 and 64Cu-D10 was accompanied by their higher uptake in other organs, particularly kidneys (Table 1 and 2). Being the major clearance organ for all the probes, kidneys showed the highest uptake with respect to other organs at every time point. Among the three probes (64Cu-M10, 64Cu-(M10)2 and64Cu-D10), the monovalent 64Cu-M10 showed the lowest kidney uptake (6.83 ± 0.35 %ID/g, 6.23 ± 0.32 %ID/g, and 3.50 ± 0.36 %ID/g at 1 h, 4 h, and 24 h p.i., respectively), while an approximate 6-fold increase of kidney uptake was observed for both divalent probes (64Cu-(M10)2: 53.6 ± 8.68 %ID/g, 46.25 ± 10.39 %ID/g, and 27.33 ± 7.3 %ID/g at 1 h, 4 h, and 24 h p.i., respectively; 64Cu-D10: 43.46 ± 1.8 %ID/g, 39.3 ± 5.66 %ID/g, and 21.36 ± 0.57 %ID/g at 1 h, 4 h, and 24 h p.i., respectively) (Figure 3 and Table 1).
Peptide Acetylation
Acetylation of the N-terminus of the H2009.1-10mer peptide was employed to protect the peptide from serum peptidases (36). This is particularly important as the critical RGD lies directly at the N-terminus of the peptide. Although acetylation did not affect serum stability in rat serum, it may protect the peptide in vivo. Additionally, acetylation reduces the charge of each peptide branch by +1 which may reduce nonspecific binding of the peptide and improve the in vivo biodistribution characteristics. The acetylated divalent probe, 64Cu-AcD10, was produced by capping the N-terminus of H2009.1-10mer peptide. Tumor accumulation of the acetylated probe 64Cu-AcD10 was almost identical to the non-acetylated dimeric probes (64Cu-(M10)2 and 64Cu-D10) (Table 1). Impressively, 64Cu-AcD10 showed a drastic reduced kidney uptake level as compared to the non-acetylated 64Cu-D10 and 64Cu-(M10)2 (Figure 3). Remarkably, it also displayed a much more favorable biodistribution profile than the non-acetylated dimers (Table 1). Of particular importance, the low background uptake of 64Cu-AcD10 in lung (1.33 ± 0.23 %ID/g, 0.65 ± 0.03 %ID/g, and 0.34 ± 0.08 %ID/g at 1 h, 4 h, and 24 h p.i., respectively) and liver (1.08 ± 0.11 %ID/g, 0.72 ± 0.09 %ID/g, and 0.55 ± 0.10 %ID/g at 1 h, 4 h, and 24 h p.i., respectively) is the highly desirable feature for NSCLC imaging. Not only was liver uptake of 64Cu-AcD10 significantly lower than the liver accumulation of the non-acetylated dimers (p < 0.01), in fact it was also at the same level as or less than that was observed for monomeric 64Cu-(M10)2 (Table 1). The 64Cu-AcD10 showed highest tumor-to-lung contrast among all the probes (Table 1). In sum, 64Cu-AcD10 exhibits superior biodistribution properties compared to the other probes.
Integrin Specificity
To assure that the probes are specific for αvβ6, the four probes were tested in animals bearing xenograft tumors which do not express αvβ6 (8). The large cell lung cancer cell line H460 was employed as this cell line does not express αvβ6 as determined by western immunoblotting, flow cytometry, and immunocytochemistry (Figure 4). In addition, no specific staining of αvβ6 was observed in a H460 tumor harvested from the xenograft model. By comparison, strong staining is observed on a similarly treated H2009 tumor. Consistent with these observations, the fluorescently labeled dimeric H2009.1-10mer peptide binds H2009 cells but not H460 cells as determined by flow cytometry over a concentration range of 0.10-200 nM. Binding was undetectable on H460 cells even at concentrations as high as 200 nM (Figure 6A). These data support the use of H460 NSCLC tumors an appropriate αvβ6-negative control.
Figure 3
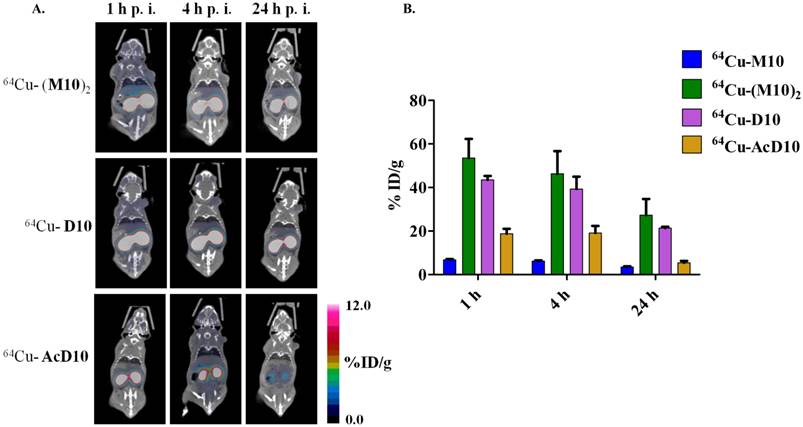
(A) Decay-corrected whole-body coronal microPET images of 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10 in SCID mice bearing H2009 tumor at 1 h, 4 h, and 24 h p.i. The kidney uptake was clearly alleviated by the peptide's N-terminus acetylation. (B) Comparative kidney uptake (%ID/g) of 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10 at 1 h, 4 h, and 24 h p.i. in H2009 tumor bearing SCID mice. Data are presented as %ID/g ± s.d. (n = 3).
Figure 4
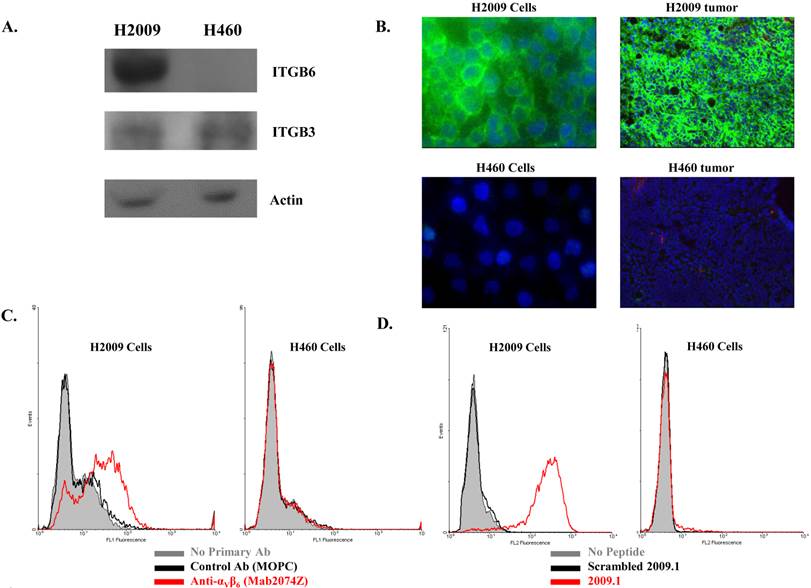
The integrin αvβ6 is expressed in H2009 cells but not in H460 cells. (A) Immunoblotting detects the β6 subunit in H2009 cells but not H460 cells (Rabbit polyclonal β6-antibody H00003694, Novus). Although weak, the β3 integrin is detected in both cell lines (Mouse monoclonal β3-antibody Ab716, Abcam). Lysate from 200,000 cells was loaded in each well. (B) Fluorescent microscopy detects expression of β6 on the surface of H2009 cells but not H460 cells (Mab2074Z). This expression is maintained in tumors grown in animals. (C) Expression of β6 on the surface of H2009 cells but not H460 cells as determined by flow cytometry. D. H2009 cells bind the H2009.1 peptide whereas the H460 cells do not, indicating the specificity of the peptide.
Consistent with the H2009.1-10mer binding αvβ6, the radiotracers showed significantly lower (p < 0.01) uptake in H460 tumor at all three time points compared to the H2009 tumors (Figure 5, Table 2). Furthermore, the ratio of H460 tumor-to-lung and tumor-to-heart ratios are ≤ 1 at 24 hours for all probes. Most importantly, individual probes showed similar biodistribution profile in H2009 and H460 tumor bearing mice (Tables 1 and 2). These data demonstrate the αvβ6-integrin specificity of the probes, 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10. Probe uptake is not a general phenomenon of all tumors but requires expression of αvβ6.
One concern is the potential for our RGD containing peptide to bind the more common αvβ3 integrin, which is a marker for neovasculature and is found on virtually all tumor vasculature. While αvβ6 is expressed specifically in H2009 tumors, αvβ3 is expressed in both H2009 and H460 tumors (Figure 4A) and tumor vasculature. Although our previous data suggests that this peptide is specific for αvβ6, we determined the binding affinity of the H2009.1-10mer peptide on purified αvβ6, αvβ3, and αvβ5 (Figure 6). The dimeric H2009.1-10mer peptide has a Kd of 0.65 ± 0.26 nM for αvβ6, 100 ± 8.5 nM for αvβ3, and 133 nM for αvβ5. This represents a 160-fold preference for αvβ6. As a reference point, the maximum concentration of the imaging probe in the blood of a mouse is ~5 nM; this is substantially below the Kd of the H2009.1-10mer dimeric peptide for αvβ3, and αvβ5 and no binding of the peptide to these purified integrins was detected at this concentration. In the same assay, the well characterized αvβ3-binding peptide cRGDyK was determined to have a Kd of 4.9 ± 2.1 nM for αvβ3 but no detectable binding to αvβ6 was observed up to 1 µM.
Table 2
Uptake 64Cu-M10, 64Cu-D10, 64Cu-AcD10 and 64Cu-(M10)2 at 1 h, 4 h and 24 h p.i. in major organs and tumor of H460 tumor bearing mice determined by quantitative PET imaging analysis. Data are presented as %ID/g ± s.d. (n = 3).
| 64Cu-M10 | 64Cu-(M10)2 | |||||
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | |
| Tumor | 0.34 ± 0.05 | 0.24 ± 0.01 | 0.19 ± 0.01 | 0.71 ± 0.05 | 0.83 ± 0.04 | 0.53 ± 0.17 |
| Kidney | 14.03 ± 2.40 | 12.50 ± 2.33 | 5.97 ± 1.01 | 85.4 ± 17.47 | 92.9 ± 11.31 | 68.9 ± 8.13 |
| Heart | 1.02 ± 0.17 | 0.65 ± 0.12 | 0.33 ± 0.03 | 1.70 ± 0.00 | 1.25 ± 0.07 | 0.83 ± 0.17 |
| Liver | 0.93 ± 0.12 | 0.77 ± 0.06 | 0.43 ± 0.05 | 5.45 ± 0.49 | 4.80 ± 0.14 | 3.25 ± 0.21 |
| Lung | 0.55 ± 0.09 | 0.34 ± 0.07 | 0.18 ± 0.02 | 1.50 ± 0.14 | 1.15 ± 0.07 | 0.77 ± 0.01 |
| Muscle | 0.15 ± 0.07 | 0.11 ± 0.04 | 0.10 ± 0.03 | 0.45 ± 0.18 | 0.31 ± 0.06 | 0.38 ± 0.08 |
| 64Cu-D10 | 64Cu-AcD10 | |||||
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | |
| Tumor | 0.86 ± 0.21 | 0.52 ± 0.05 | 0.39 ± 0.04 | 0.73 ± 0.13 | 0.45 ± 0.11 | 0.35 ± 0.05 |
| Kidney | 43.3 ± 5.4 | 50.13 ± 3.87 | 19.83 ± 2.38 | 19.5 ± 0.36 | 18.43 ± 0.92 | 3.43 ± 1.8 |
| Heart | 1.41 ± 0.43 | 0.77 ± 0.09 | 0.47 ± 0.09 | 0.82 ± 0.05 | 0.50 ± 0.04 | 0.26 ± 0.0 |
| Liver | 2.63 ± 0.92 | 2.3 ± 0.26 | 1.83 ± 0.15 | 1.13 ± 0.15 | 0.95 ± 0.06 | 0.71 ± 0.04 |
| Lung | 1.33 ± 0.30 | 0.77 ± 0.07 | 0.45 ± 0.17 | 1.02 ± 0.06 | 0.65 ± 0.02 | 0.24 ± 0.10 |
| Muscle | 0.75 ± 0.24 | 0.31 ± 0.06 | 0.33 ± 0.08 | 0.61 ± 0.12 | 0.35 ± 0.05 | 0.19 ± 0.13 |
Figure 5
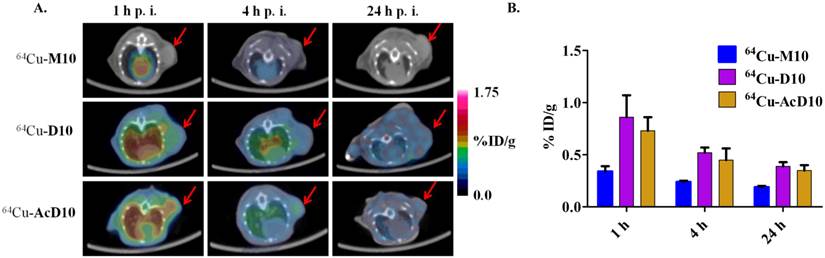
(A) Decay-corrected trans-axial microPET images of 64Cu-M10, 64Cu-D10, and 64Cu-AcD10 in SCID mice bearing H460 tumor from static scan at 1 h, 4 h, and 24 h p.i. Tumors are indicated by red arrow. (B) Comparative tumor uptakes (%ID/g) of 64Cu-M10, 64Cu-D10, and 64Cu-AcD10 at 1 h, 4 h, and 24 h p.i. Data are presented as %ID/g ± s.d. (n = 3).
Figure 6
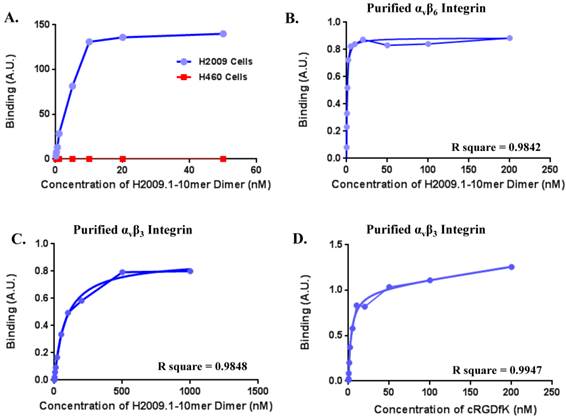
(A)The dimeric H2009.1 peptide binds specifically to αvβ6-expressing H2009 cells but not to αvβ6-negative H460 cells. Cells were incubated with the indicated concentration of labeled dimeric peptide for 1 h at 37oC. Peptide uptake in cells was determined by flow cytometry. The arbitrary binding units reflect the mean fluorescence intensity. (B-D)Representative binding curves are shown for the H2009.1 dimeric peptide on purified αvβ6 (B) and αvβ3 (C). Control cRDGDfK peptide was used to assure the activity of the αvβ3 protein (D). Dissociation constants were determined using nonlinear regression in GraphPad Prism using assuming a one-site binding model. All experiments were repeated 3-times.
We further evaluated the specific αvβ3 integrin expression in H2009 and H460 tumors using our previously reported peptide-RGD based dimeric probe (64Cu-CB-TE2A-(c(RGDyK))2) (Figure 7 and Table 3). Unlike 64Cu-M10, 64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10, 64Cu-CB-TE2A-(c(RGDyK))2 clearly visualized both tumors and showed nearly identical tumor uptake at all three time points (1 h: 1.58 ± 0.13 %ID/g in H2009 vs 1.64 ± 0.20 %ID/g in H460; 4 h: 1.07 ± 0.21 %ID/g in H2009 vs 1.09 ± 0.24 %ID/g in H460; 24 h: 0.70 ± 0.29 %ID/g in H2009 vs. 0.72 ± 0.10 %ID/g in H460). In sum, our data confirms that four probes built upon the H2009.1-10mer peptide do not recognize αvβ3 but specifically bind to αvβ6 in NSCLCs. Of note, 64Cu-CB-TE2A-(c(RGDyK))2 exhibited significantly lower kidney uptake in both tumor models than 64Cu-(M10)2, 64Cu-D10, or 64Cu-AcD10 (p < 0.0001), which indicates that the CB-TE2A chelator scaffold has a negligible role in the high kidney accumulation.
Discussion
The role of molecular tumor profiling is ever-increasing in clinical practice for diagnosis, treatment planning and monitoring of disease. There is an increasing demand to diagnose tumors based on prognostic biomarkers in order to select the therapy that is most likely to successfully treat an individual patient's cancer. The αvβ6-integrin is an emerging prognostic biomarker for NSCLC as well as other epithelial based cancer. Expression of αvβ6 in NSCLC patients is associated with poor survival. The up-regulation of αvβ6-integrin in early stage NSCLC, as well as its association with a pro-invasive NSCLC phenotype, makes it highly desirable target for diagnostic and therapeutic application (11, 12). To date, a few αvβ6-targeted probes have been reported. Of which, most efforts are based on a 20 amino acid peptide derived from the coat protein of foot and mouth disease virus (A20FMDV2, Sequence: NAVPNLRGDLQVLAQKVART). This peptide sequence binds to αvβ6 with low nanomolar affinity. When modified and labeled with 18F, the peptidic PET probe ([18F]FBA-A20FMDV2) was able to show a reasonable tumor uptake (0.66 %ID/g at 1 h) in an αvβ6+ tumor model, although the tumor uptake retention needs to be further improved (37, 38).
Recently, a αvβ6-binding cysteine knot was engineered to improve in vivo stability (39, 40). This ligand displays similar affinity and specificity for αvβ6 as the A20FMDV peptide. When modified and labeled with 64Cu, the cysteine knot showed good tumor uptake and retention (1.8 ± 0.50%, 1 h and 1.46 ± 0.43%, 24 h). However, the modest tumor to lung ratio (1.89 ±0.81%, 1 h and 1.25 ± 0.28%, 24 h) may hamper the use of this probe for detection of NSCLC tumors. Additionally, this probe requires biosynthesis and may invoke an immunological response upon repeat injections.
Figure 7
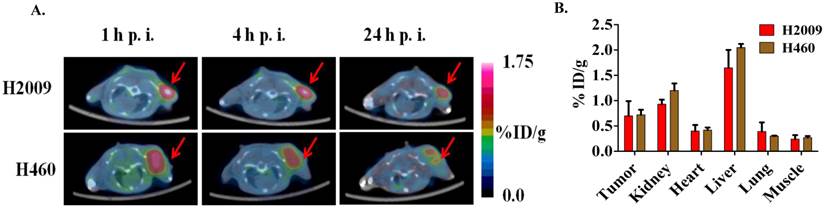
(A) Decay-corrected trans-axial microPET images of 64Cu-CB-TE2A-(c(RGDyK))2 in SCID mice bearing H2009 and H460 tumors at 1 h, 4 h, and 24 h p.i.. Tumors are indicated by red arrow. (B) Comparative uptake of 64Cu-CB-TE2A-(c(RGDyK))2 in H2009 and H460 tumors at 24 h p.i. Data are presented as %ID/g ± s.d. (n = 3).
Table 3
Uptake 64Cu-CB-TE2A-(c(RGDyK))2 at 24 h p.i. in major organs and tumor of H2009 and H460 tumor bearing mice determined by quantitative PET imaging analysis. Data are presented as %ID/g ± s.d. (n = 3).
| H2009 Tumor | H460 Tumor | |||||||
|---|---|---|---|---|---|---|---|---|
| 1 h | 4 h | 24 h | 1 h | 4 h | 24 h | |||
| Tumor | 1.58 ± 0.13 | 1.07 ± 0.21 | 0.70 ± 0.29 | 1.64 ± 0.20 | 1.09 ± 0.24 | 0.72 ± 0.10 | ||
| Kidney | 3.43 ± 0.25 | 1.35 ± 0.54 | 0.93 ± 0.09 | 4.33 ± 1.0 | 1.80 ± 0.57 | 1.2 ± 0.14 | ||
| Heart | 1.67 ± 0.13 | 0.58 ± 0.13 | 0.40 ± 0.12 | 1.90 ± 0.72 | 0.54 ± 0.26 | 0.42 ± 0.05 | ||
| Liver | 2.56 ± 0.41 | 1.97 ± 0.19 | 1.65 ± 0.35 | 2.13 ± 0.25 | 1.61 ± 0.41 | 2.05 ± 0.07 | ||
| Lung | 0.70 ± 0.05 | 0.61 ± 0.17 | 0.39 ± 0.18 | 0.68 ± 0.02 | 0.48 ± 0.06 | 0.30 ± 0.01 | ||
| Muscle | 0.41 ± 0.04 | 0.26 ± 0.12 | 0.24 ± 0.08 | 0.39 ± 0.06 | 0.31 ± 0.04 | 0.27 ± 0.03 | ||
Success of a αvβ6-specific PET imaging probe depends upon the choice of a ligand with high binding affinity and selectivity towards αvβ6-integrin. The H2009.1 peptide was selected by biopanning a phage displayed-peptide library on intact H2009 NSCLC cells. Subsequently, we determined that the H2009.1 peptide binds with high binding affinity and selectivity to αvβ6+ (8, 20). Not surprisingly, the selected peptide sequence contains an Arginine-Glycine-Aspartate (RGD) tripeptide sequence, a well-known ligand for many integrins (41, 42). However, the peptide is specific for αvβ6 and does not bind other RGD-binding integrins (8). Importantly, this peptide does not bind cells that express other RGD binding integrins including αvβ5, αvβ3 and α5β1 but lack αvβ6. Furthermore, we have shown here that the H2009.1-10mer dimeric peptide has a >160-fold preference for purified αvβ6 for compared to αvβ5 and αvβ3. The specificity of the H2009.1 peptide for αvβ6-integrin arises from the peptide sequence LATL directly to the carboxy-terminal side of the RGD. The sequence LXXL has been shown to impart αvβ6-specificity when it flanks the RGD domain (18, 43-46). Notably, the binding of H2009.1 peptide showed a linear correlation with the αvβ6-integrin expression on the cell surface, which enables the H2009.1 peptide as an ideal ligand for αvβ6+ tumor detection as well as quantification (8, 20). However, like other peptides selected from phage displayed libraries, the corresponding synthetic monomer peptide was not ideal for molecular imaging, especially at the later time points, primarily due to suboptimal binding affinity (47). The phage-displayed library, from which the H2009.1 peptide was selected, presents the peptide at the N-terminus of the phage pIII coat protein. As such, the peptide is displayed at one end of the filamentous phage in 3 - 5 copies. Thus, target-specific binding of the phage is likely driven by both the peptide sequence and multimeric presentation of peptides.
Multimeric presentation of the H2009.1-10mer peptide was sought to increase the receptor-ligand binding affinity while maintaining the specificity of the peptide for tumor detection with the aim to enhance the specific PET imaging signal (22, 26, 29). Given that juxtaposition positioning of H2009.1-10mer peptide in the three dimensional space might play a key role in ligand-receptor interaction on the cell surface, we employed two complimentary multimerization approaches to evaluate the anticipated multivalent effect. First, simultaneous presentation of two monomeric peptides on a divalent BFC scaffold (CB-TE2A(tBu)2-(Mal)2) gave a scaffold-based divalent conjugate H2(M10)2. Second, the isolated H2009.1-10mer peptide was dimerized and subsequently presented on a monovalent BFC scaffold (CB-TE2A(tBu)2-Mal) to provide peptide-based divalent conjugates H2D10 and H2(Ac)D10. In all cases, a polyethylene glycol (PEG) linker was included in the peptide design in order to increase the solubility of the resulted peptide conjugates in aqueous solution and to act as a spacer between the chelating scaffold and the peptide moiety for optimal in vivo kinetics and high tumor uptake. Indeed, it was reported that attaching a defined length 28-PEG spacer to A20FMDV2 could provide a 3-fold tumor uptake increase, although higher uptake was also observed in non-target organs (37).
The multivalent effect on binding affinity of isolated peptides was evaluated in a cell-based assay using H2009 human NSCLC cell line. As expected, divalent conjugates of H2009.1-10mer peptide (H2-(M10)2, H2-D10, and H2-AcD10) showed a 4 - 6 fold enhancement in their binding affinity over the monovalent one (H2-M10). The greater than 2-fold increase in affinity is indicative of multivalent binding of the peptide to the cell surface and not merely an increase in the number of peptide units. Attachment of the peptides to the CB-TE2A chelator does not affect the affinity of the peptide compared to the parental dimer of H2009.1-10mer. Interestingly, the two divalent conjugates, H2-(M10)2 and H2-D10, have the same affinity for H2009 cells. Thus displaying the peptide off a lysine core or the divalent CB-TE2A core does not significantly affect the specific cell binding. Of note, CB-TE2A conjugates of the A20FMDV peptide have also been reported (48). However, in the modification one of the carboxylate groups was consumed for the peptide conjugation, which might compromise the stability of the Cu(II) complex moiety. In addition, it adds an extra positive charge to the peptide conjugate when labeled with 64Cu, which might contribute to the high kidney uptake. In contrast, all our peptide conjugates maintain an intact CB-TE2A core to form the neutral Cu(II)-CB-TE2A complex with non-compromised stability.
Towards the goal of utilizing the multivalent effect for αvβ6-integrin PET imaging, we conducted non-invasive imaging evaluation studies in two SCID mouse models bearing H2009 (αvβ6+) and H460 (αvβ6-) tumors. The H2009 (αvβ6+) tumor was clearly visualized by all four probes, monovalent (64Cu-M10) and divalent (64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10), at 1 h p.i. In contrast, much lower tumor uptake was observed for all the probes in H460 (αvβ6-) tumor while their distribution profiles in the other tissue were almost identical to those observed in the H2009 tumor-bearing mice. This result demonstrates the αvβ6 binding specificity of the H2009.1-10mer peptide. However, though the H2009 tumor was visualized by 64Cu-M10 at 1 h p.i., its imaging contrast became barely visible at 4 and 24 h p.i. The low tumor contrast of 64Cu-M10 is likely caused by its suboptimal in vivo kinetics, receptor binding, and stability. These drawbacks can be potentially resolved or mitigated by multimeric presentation of the peptide on a scaffold. Indeed, the divalent probes (64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10) showed an approximate 3-fold tumor uptake increase as compared to 64Cu-M10, which clearly demonstrates the role of the multivalent effect in imaging signal amplification. In addition, the tumor PET signal retention of the three divalent probes stayed above 0.70 %ID/g even at 24 h p.i. While the increased affinity of the dimeric probes contributes to the increased tumor accumulation, increased stability and improved in vivo kinetics are likely to play a role as well. Interestingly, no significant tumor uptake difference was observed for the three divalent probes out to 24 h p.i., indicating the two multivalent presentation approaches afforded virtually the same result. While this observation was somewhat to our surprise, it may result from the flexible PEG spacer that has been incorporated into our probe design, which provides no spatial constraints to the ligand-receptor binding. All three divalent probes showed a similar biodistribution profile in the H2009 and H460 tumor models with elevated uptake (< 3 fold) in major organs other than kidneys as compared to the monovalent one. This is likely caused by the molecular weight induced difference of in vivo kinetics.
Although we observe relatively high tumor uptake compared to background accumulation in the mediastinum region, we recognize the need to further increase accumulation of radiolabel in the tumor. Improved tumor uptake is anticipated to increase sensitivity of detection. One advantage of our bifunctional chelator design is that we can layer the valency, moving rapidly from monomeric to octomeric peptides. As the valency increases, we anticipate an increase in affinity for αvβ6 and improved in vivo kinetics (30). Additionally, the design allows for facile PEGylation of both the peptide ligand and the chelator which may improve tumor uptake by increasing circulation time of the probe (49). Defined length PEG linkers are available allowing for controlled, empirical testing.
Rapid kidney uptake and efficient clearance is a desired feature of targeted PET imaging probes. However, high kidney uptake is unwanted even for lung cancer imaging because it renders the kidneys to unnecessary radiation exposure. Given the high kidney uptake and retention observed for both 64Cu-(M10)2 and 64Cu-D10 (Figure 3, Table 1), they may not find practical applications in PET imaging of αvβ6-integrin. By immunocytochemical staining, we found negligible expression of αvβ6 integrin in the kidneys of H2009 tumor bearing mice (data not shown), indicating the non-specific nature of the observed high kidney accumulation. Further, as shown in the PET imaging results of 64Cu-CB-TE2A-(c(RGDyK))2 in both H2009 and H460 tumor models, the CB-TE2A-based construct is not the culprit of the high kidney uptake. Therefore we reasoned that the high kidney uptake and retention resulted from the peptide itself. Two factors may play an important role - the peptide's stability and charge in vivo.
As such, we introduced an acetyl group to cap the N-terminus of the H2D10 conjugate, which led to H2AcD10. The acetylation reduces the overall charge from +4 (H2D10) to +2 (H2AcD10) while limiting proteolysis of the peptide by N-terminal peptidases (36). As anticipated, 64Cu-AcD10 showed a drastic decrease in kidney uptake and retention and the level of tumor uptake was maintained compared to its non-acetylated counterpart, 64Cu-D10 (Figure 3). In addition, a significant uptake reduction was also observed in other non-target organs, including lung and liver. The improved tumor-to-lung contrast is of great importance to the imaging detection of NSCLCs. These observations clearly demonstrate that acetylation of peptide's N-terminus is an efficient way to reduce the nonspecific kidney accumulation and optimize the in vivo kinetics of peptide-based imaging probes. To our surprise, the apparent stability profile of all three divalent probes (64Cu-(M10)2, 64Cu-D10, and 64Cu-AcD10) measured in rat serum out to 24 h were almost identical. This indicates that the in vivo properties of the divalent probe were improved as the result of its positive charge reduction. However, we cannot rule out the possibility that acetylation improves the in vivo stability in mice. It is important to note that the previous PET studies with A20FMDV2 also used a capped N-terminus and the kidney uptake varied greatly. Similarly, the αvβ6-binding cysteine knot probes showed persistent kidney retention. Therefore, simple capping of the N-terminus does not abrogate kidney retention for all αvβ6 PET probes. Additionally, it should be noted that multimerization of other peptidic ligands has also lead to a significant increase in kidney accumulation (30, 48).
Kidney retention of peptidic probes is a complicated, multifactorial issue and involves numerous factors including peptide stability, charge, hydrophobicity, choice of radiolabel, peptide sequence, and chemical modifications of the peptide (50-52). Due to their size, peptides are filtered by glomeruli and excreted; however, a small percentage of the peptide or peptide fragments are reabsorbed by the proximal tubules and retained. Although unlikely to induce substantial renal toxicity within the context of imaging, retention of the radiolabel can affect detection of tumors within or around the kidneys. However, kidney accumulation can be problematic for therapeutic applications in which the peptide is used to deliver radiopharmaceuticals or chemotherapeutics. As such efforts to reduce peptidic retention in the kidneys is of key importance. Minor changes in the charge and/or chemical structure of a peptide have been shown to dramatically affect renal uptake. Pre-dosing animals with polycationic species or Gelofusine prior to administering radiolabeled peptides has been observed to reduce kidney retention. PEGylation of peptides has also been shown to improve tumor-to-kidney uptake ratio. Although we have shown significant reduction of kidney retention by acetylating the peptide, we are performing further empirical studies to minimize kidney accumulation of the radiolabeled peptidic probes.
Integrins are commonly found in tumor cells as well as in angiogenic tumor vasculature. As previously mentioned, the H2009.1-10mer peptide sequence contains an RGD moiety, which is an overlapping ligand for various integrins such as αvβ3 integrin. Although, in vitro data support the specificity of the H2009-10mer peptide for αvβ6, one may question whether our imaging results indeed reflect the αvβ6 expression in tumor. To answer this question, we performed an imaging study in the same tumor models using our recently reported conjugate (64Cu-CB-TE2A-(c(RGDyK))2) built on the same BFCS, which specifically targets the αvβ3 integrin. The cRGDyX peptide is widely used as an αvβ3-specific ligand for both molecular imaging of angiogenic tumor vasculature and anti-angiogenic therapies (53). Unlike H2009.1-10mer conjugates, the 64Cu-CB-TE2A-(c(RGDyK))2 showed virtually identical uptake in H2009 and H460 tumors out to 24 h p.i. (1.8 %ID/g at 1h p.i.; 0.7 %ID/g at 24 h p.i.). Lack of specificity of 64Cu-CB-TE2A-(c(RGDyK))2 can be attributed to ubiquitous expression of αvβ3 integrin in angiogenic vessels as well as in tumor cells. Unlike 64Cu-CB-TE2A-(c(RGDyK))2, the specific imaging of H2009 tumor by 64Cu-(M10)2, 64Cu-D10, or 64Cu-AcD10 is primarily due to the restrictive expression of αvβ6 integrin in H2009 tumor. These results clearly demonstrate the potential use of our designed probes for molecular profiling of αvβ6+ NSCLC.
Conclusions
Imparting multivalency is an effective way to improve biopotency of a phage selected peptide. The designed multivalent probes showed enhanced binding affinity, which was utilized for PET signal enhancement in αvβ6+ tumor imaging. Significantly, multivalent probes maintained the desired specificity to image αvβ6+ H2009 tumor, while low signal was observed in αvβ6- H460 tumor. We showed that N-terminus acetylated probe (64Cu- AcD10) provided drastic uptake reduction in kidney and other non-target organs while maintaining tumor uptake. Further evaluation of this methodology is under way to realize its full potential in imaging probe design by utilizing existing ligands selected from combinatorial library screening. Overall, the selective tumor uptake of 64Cu-AcD10 along with its favorable distribution in major organs makes it an ideal candidate to be developed for specific imaging of αvβ6+ expression. Additionally, 67Cu can be used for radiotherapy, opening the possibility of using this probe as a therapeutic as well.
Acknowledgements
This work was partially supported by a small animal imaging research program grant (SAIRP) from the National Institute of Cancer (U24 CA126608 XS), the Welch Foundation (I-1622 KCB) and the National Institute of Biomedical Imaging and Bioengineering (1R01EB014244-01 XS and KCB). The authors acknowledge the generous support of a private donor that allowed the purchase of the Siemens Inveon PET-CT Multi-modality System. We thank Bethany Powell Gray for obtaining the data found in Figure 4B.
Conflict of Interests
The authors declare no conflicts of interest.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: Cancer J Clin. 2013;63:11-30
2. Janku F, Stewart DJ, Kurzrock R. Targeted therapy in non-small-cell lung cancer-Is it becoming a reality? Nat Rev Clin Oncol. 2010;7:401-14
3. Ludwig JA, Weinstein JN. Biomarkers in cancer staging, prognosis and treatment selection. Nat Rev Cancer. 2005;5:845-56
4. Silvestri GA, Gould MK, Margolis ML, Tanoue LT, Mccrory D, Toloza E. et al. Noninvasive staging of non-small cell lung cancer - ACCP evidenced-based clinical practice guidelines. Chest. 2007;132:178s-201s
5. Harry VN, Semple SI, Parkin DE, Gilbert FJ. Use of new imaging techniques to predict tumour response to therapy. Lancet Oncol. 2010;11:92-102
6. Phelps ME. Positron emission tomography provides molecular imaging of biological processes. Proc Natl Acad Sci USA. 2000;97:9226-33
7. Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY. et al. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11:2785-808
8. Elayadi AN, Samli KN, Prudkin L, Liu Y-H, Bian A, Xie X-J. et al. A peptide selected by biopanning identifies the integrin αvβ6 as a prognostic biomarker for nonsmall cell lung cancer. Cancer Res. 2007;67:5889-95
9. Breuss JM, Gillett N, Lu L, Sheppard D, Pytela R. Restricted distribution of integrin ß6 mRNA in primate epithelial tissues. J Histochem Cytochem. 1993;41:1521-7
10. Bates RC, Bellovin DI, Brown C, Maynard E, Wu B, Kawakatsu H. et al. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 2005;115:339-47
11. Prudkin L, Liu DD, Ozburn NC, Sun M, Behrens C, Tang X. et al. Epithelial-to-mesenchymal transition in the development and progression of adenocarcinoma and squamous cell carcinoma of the lung. Mod Path. 2009;22:668-78
12. Ramos DM, Dang D, Sadler S. The role of the integrin αvβ6 in regulating the epithelial to mesenchymal transition in oral cancer. Anticancer Res. 2009;29:125-30
13. Ahmed N, Riley C, Rice GE, Quinn MA, Baker MS. αvβ6 integrin-A marker for the malignant potential of epithelial ovarian cancer. J Histochem Cytochem. 2002;50:1371-9
14. Hazelbag S, Kenter GG, Gorter A, Dreef EJ, Koopman LA, Violette SM. et al. Overexpression of the αvβ6 integrin in cervical squamous cell carcinoma is a prognostic factor for decreased survival. J Pathol. 2007;212:316-24
15. Thomas GJ, Lewis MP, Whawell SA, Russell A, Sheppard D, Hart IR. et al. Expression of the αvβ6 integrin promotes migration and invasion in squamous carcinoma cells. J Invest Derm. 2001;117:67-73
16. Yang GY, Xu KS, Pan ZQ, Zhang ZY, Mi YT, Wang JS. et al. Integrin αvβ6 mediates the potential for colon cancer cells to colonize in and metastasize to the liver. Cancer Sci. 2008;99:879-87
17. Goodman SL, Hoelzemann G, Sulyok GaG, Kessler H. Nanomolar small molecule inhibitors for αvβ6, αvβ5, and αvβ3 Integrins. J Med Chem. 2002;45:1045-51
18. Dicara D, Rapisarda C, Sutcliffe JL, Violette SM, Weinreb PH, Hart IR. et al. Structure-function analysis of Arg-Gly-Asp helix motifs in αvβ6 integrin ligands. J Biol Chem. 2007;282:9657-65
19. Kraft S, Diefenbach B, Mehta R, Jonczyk A, Luckenbach GA, Goodman SL. Definition of an unexpected ligand recognition motif for αvβ6 integrin. J Biol Chem. 1999;274:1979-85
20. Li S, Mcguire MJ, Lin M, Liu Y-H, Oyama T, Sun X. et al. Synthesis and characterization of a high-affinity αvβ6-specific ligand for in vitro and in vivo applications. Mol Cancer Ther. 2009;8:1239-49
21. Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed Engl. 2006;45:2348-68
22. Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed Engl. 1998;37:2755-94
23. Bracci L, Falciani C, Lelli B, Lozzi L, Runci Y, Pini A. et al. Synthetic peptides in the form of dendrimers become resistant to protease activity. J Biol Chem. 2003;278:46590-5
24. Falciani C, Lozzi L, Pini A, Corti F, Fabbrini M, Bernini A. et al. Molecular basis of branched peptides resistance to enzyme proteolysis. Chem Biol Drug Des. 2007;69:216-21
25. Liu W, Hao GY, Long MA, Anthony T, Hsieh JT, Sun X. Imparting multivalency to a bifunctional chelator: A scaffold design for targeted PET imaging probes. Angew Chem Int Ed Engl. 2009;48:7346-9
26. Singh AN, Liu W, Hao GY, Kumar A, Gupta A, Oz OK. et al. Multivalent bifunctional chelator scaffolds for Gallium-68 based positron emission tomography imaging probe design: Signal amplification via multivalency. Bioconj Chem. 2011;22:1650-62
27. Dijkgraaf I, Kruijtzer JaW, Liu S, Soede AC, Oyen WJG, Corstens FHM. et al. Improved targeting of the alpha(v)beta(3) integrin by multimerisation of RGD peptides. Eur J Nucl Med Mol Imaging. 2007;34:267-73
28. Janssen M, Oyen WJG, Massuger LFaG, Frielink C, Dijkgraaf I, Edwards DS. et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm. 2002;17:641-6
29. Liu S. Radiolabeled cyclic RGD peptides as integrin αvβ6-targeted radiotracers: Maximizing binding affinity via bivalency. Bioconj Chem. 2009;20:2199-213
30. Li Z-B, Cai W, Cao Q, Chen K, Wu Z, He L. et al. 64Cu-Labeled tetrameric and octameric RGD peptides for small-animal PET of tumor αvβ3 integrin expression. J Nuc Med. 2007;48:1162-71
31. Wong EH, Weisman GR, Hill DC, Reed DP, Rogers ME, Condon JS. et al. Synthesis and characterization of cross-bridged cyclams and pendant-armed derivatives and structural studies of their copper(II) complexes. J Am Chem Soc. 2000;122:10561-72
32. Phelps RM, Johnson BE, Ihde DC, Gazdar AF, Carbone DP, Linnoila P. et al. NCI-Navy medical oncology branch cell line database. J Cell Biochem. 1996(Suppl 24):32-91
33. Jensen M, Jorgensen J, Binderup T, Kjaer A. Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Medical Imaging. 2008;8:16
34. Mcguire MJ, Li S, Brown KC. Biopanning of phage displayed peptide libraries for the isolation of cell-specific ligands. In: Rasooly A, Herold K, editors. Biosensors and Biodetectors Methods and Protocols Volume 504: Electochemical and Mechanical Detectors, Lateral Flow, and Ligands for Biosensors. Methods in Molecular Biology. New York, New York: Humana Press. 2009;504:291-321
35. Benuck M, Marks N. Differences in the degradation of hypothalamic releasing factors by rat and human serum. Life Sci. 1976;19:1271-6
36. Werle M, Bernkop-Schnürch A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids. 2006;30:351-67
37. Hausner SH, Abbey CK, Bold RJ, Gagnon MK, Marik J, Marshall JF. et al. Targeted in vivo Imaging of integrin αvβ6 with an improved radiotracer and its relevance in a pancreatic tumor model. Cancer Res. 2009;69:5843-50
38. Hausner SH, Dicara D, Marik J, Marshall JF, Sutcliffe JL. Use of a peptide derived from foot-and-mouth disease virus for the noninvasive imaging of human cancer: Generation and evaluation of 4-[18F]fluorobenzoyl A20FMDV2 for in vivo imaging of integrin αvβ6 expression with positron emission tomography. Cancer Res. 2007;67:7833-40
39. Kimura RH, Teed R, Hackel BJ, Pysz MA, Chuang CZ, Sathirachinda A. et al. Pharmacokinetically stabilized cystine knot peptides that bind αvβ6 integrin with single-digit nanomolar affinities for detection of pancreatic cancer. Clin Cancer Res. 2012;18:839-49
40. Hackel BJ, Kimura RH, Miao Z, Liu H, Sathirachinda A, Cheng Z. et al. 18F-Fluorobenzoate-labeled cystine knot peptides for PET imaging of integrin αvβ6. J Nuc Med. 2013;54:1101-5
41. Avraamides CJ, Garmy-Susini B, Varner JA. Integrins in angiogenesis and lymphangiogenesis. Nat Rev Cancer. 2008;8:604-17
42. Ruoslahti E. RGD and other recognition sequences for integrins. Annu Rev Cell Dev Biol. 1996;12:697-715
43. Gagnon M, Hausner S, Marik J, Abbey C, Marshall J, Sutcliffe J. High-throughput in vivo screening of targeted molecular imaging agents. Proc Natl Acad Sci USA. 2009;106:17904-9
44. Hausner S, Abbey C, Bold R, Gagnon K, Marik J, Marshall J. et al. Targeted in vivo imaging of inetgrin αvβ6 with an improved radiotracer and its relevance in a pancreatic tumor model. Cancer Res. 2009;69:5843-50
45. Hausner S, Dicara D, Marik J, Marshall JF, Sutcliffe JL. Use of a peptide derived from foot-and-mouth disease virus for the noninvasive imaging of human cancer: Generation and evaluation of 4-[18F]fluorobenzoyl A20FMDV2 for in vivo imaging of integrin αvβ6 expression with positron emission tomography. Cancer Res. 2007;67:7833-7
46. Kraft S, Diedenbach B, Mehta R, Jonczyk A, Luckenbach GA, Goodman SL. Definition of an unexpected ligand recognition motif for αvβ6 integrin. J Biol Chem. 1999;274:1979-85
47. Bastings MMC, Helms BA, Van Baal I, Hackeng TM, Merkx M, Meijer EW. From phage display to dendrimer display: Insights into multivalent binding. J Am Chem Soc. 2011;133:6636-41
48. Hausner S, Kukis D, Gagnon K, Stanecki C, Ferdani R, Marshall J. et al. Evaluation of [64Cu]Cu-DOTA and [64Cu]Cu-CB-TE2A chelates for targeted positron emission tomography with an αvβ6-specific peptide. Molecular Imaging. 2009;8:111-21
49. Li L, Turatti F, Crow D, Bading JR, Anderson A-L, Poku E. et al. Monodispersed DOTA-PEG-conjugated anti-TAG-72 diabody has low kidney uptake and high tumor-to-blood ratios resulting in improved 64Cu PET. J Nuc Med. 2010;51:1139-46
50. Vegt E, De Jong M, Wetzels JFM, Masereeuw R, Melis M, Oyen WJG. et al. Renal toxicity of radiolabeled peptides and antibody fragments: mechanisms, impact on radionuclide therapy, and strategies for prevention. J Nuc Med. 2010;51:1049-58
51. Rolleman EJ, Melis M, Valkema R, Boerman OC, Krenning EP, De Jong M. Kidney protection during peptide receptor radionuclide therapy with somatostatin analogues. European Journal of Nuclear Medicine & Molecular Imaging. 2010;37:1018-31
52. De Jong M, Breeman W, Kwekkeboom D, Valkema R, Krenning EP. Tumor imaging and therapy using radiolabeled somatostatin analogues. Acc Chem Res. 2010;42:873-80
53. Gaertner FC, Kessler H, Wester HJ, Schwaiger M, Beer AJ. Radiolabelled RGD peptides for imaging and therapy. Eur J Nucl Med Mol Imaging. 2012;39:126-38
Author contact
![]() Corresponding author: Kathlynn C. Brown, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8807. Tel: 214-645-6348 Fax: 214-645-6347Kathlynn.Brownedu. Xiankai Sun, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8542. Tel: 214-645-5978 Fax: 214-645-2885. Xiankai.Sunedu.
Corresponding author: Kathlynn C. Brown, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8807. Tel: 214-645-6348 Fax: 214-645-6347Kathlynn.Brownedu. Xiankai Sun, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-8542. Tel: 214-645-5978 Fax: 214-645-2885. Xiankai.Sunedu.
Citation styles
APA
Singh, A.N., McGuire, M.J., Li, S., Hao, G., Kumar, A., Sun, X., Brown, K.C. (2014). Dimerization of a Phage-Display Selected Peptide for Imaging of αvβ6- Integrin: Two Approaches to the Multivalent Effect. Theranostics, 4(7), 745-760. https://doi.org/10.7150/thno.7811.
ACS
Singh, A.N.; McGuire, M.J.; Li, S.; Hao, G.; Kumar, A.; Sun, X.; Brown, K.C. Dimerization of a Phage-Display Selected Peptide for Imaging of αvβ6- Integrin: Two Approaches to the Multivalent Effect. Theranostics 2014, 4 (7), 745-760. DOI: 10.7150/thno.7811.
NLM
Singh AN, McGuire MJ, Li S, Hao G, Kumar A, Sun X, Brown KC. Dimerization of a Phage-Display Selected Peptide for Imaging of αvβ6- Integrin: Two Approaches to the Multivalent Effect. Theranostics 2014; 4(7):745-760. doi:10.7150/thno.7811. https://www.thno.org/v04p0745.htm
CSE
Singh AN, McGuire MJ, Li S, Hao G, Kumar A, Sun X, Brown KC. 2014. Dimerization of a Phage-Display Selected Peptide for Imaging of αvβ6- Integrin: Two Approaches to the Multivalent Effect. Theranostics. 4(7):745-760.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.