Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
CXCR4 structure
Role of CXCR4 in HIV Infection
Role of CXCR4 in Cancer Cell...
CXCR4 and Rheumatoid Arthritis
CXCR4 and WHIM Syndrome
CXCR4 and Ischemic Injury
CXCR4 and Pulmonary Fibrosis
CXCR4 and Systemic Lupus...
Discovery of CXCR4 Antagonists
Cyclic Pentapeptide-based...
Indole-based CXCR4 Antagonists
AMD070 Derivatives:...
Para-xylyl-enediamine-based...
Guanidine-based CXCR4 Antagonists
Quinoline derivatives
Miscellaneous Compounds
CXCR4 Antagonists in Clinical...
Conclusions
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(1):47-75. doi:10.7150/thno.5376 This issue Cite
Review
Small Molecule Inhibitors of CXCR4
Bikash Debnath1, Shili Xu1, Fedora Grande2, Antonio Garofalo2, Nouri Neamati1 ![]()
1. Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, 1985 Zonal Avenue, PSC 304, Los Angeles, CA 90089-9121, USA.
2. Dipartimento di Scienze Farmaceutiche, Università della Calabria, 87036 Arcavacata di Rende, CS Italy.
Received 2012-10-12; Accepted 2012-12-4; Published 2013-1-15
Abstract
CXCR4 is a G-protein-coupled receptor involved in a number of physiological processes in the hematopoietic and immune systems. The SDF-1/CXCR4 axis is significantly associated with several diseases, such as HIV, cancer, WHIM syndrome, rheumatoid arthritis, pulmonary fibrosis and lupus. For example, CXCR4 is one of the major co-receptors for HIV entry into target cells, while in cancer it plays an important role in tumor cell metastasis. Several promising CXCR4 antagonists have been developed to block SDF-1/CXCR4 interactions that are currently under different stages of development. The first in class CXCR4 antagonist, plerixafor, was approved by the FDA in 2008 for the mobilization of hematopoietic stem cells and several other drugs are currently in clinical trials for cancer, HIV, and WHIM syndrome. While the long-term safety data for the first generation CXCR4 antagonists are not yet available, several new compounds are under preclinical development in an attempt to provide safer and more efficient treatment options for HIV and cancer patients.
Keywords: CXCR4, antagonists, HIV, cancer, rheumatoid arthritis, WHIM syndrome, lupus.
Introduction
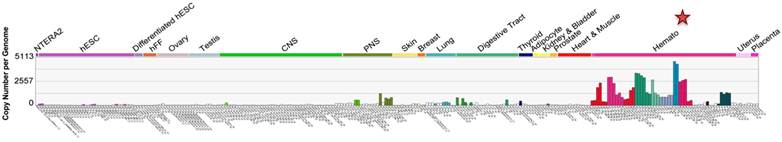
C-X-C chemokine receptor type 4 (CXCR4), also known as fusin or cluster of differentiation 184 (CD184), is a seven transmembrane (TM) G-protein coupled receptor (GPCR) belonging to Class I GPCR or rhodopsin-like GPCR family [1, 2]. Under normal physiological conditions, CXCR4 is mainly expressed in the hematopoietic and immune systems (Figure 1), where it carries out multiple functions. CXCR4 was initially discovered as one of the co-receptors involved in human immunodeficiency virus (HIV) cell entry. Subsequently, it was found to be expressed in a wide variety of tissues, including lymphatic tissues, thymus, brain, spleen, stomach and small intestine [3-5].
CXCR4 expression in embryonic and adult normal tissues (U133A). CXCR4 levels in various body tissues are displayed (image generated using Amazonia!: http://amazonia.transcriptome.eu [129]).
The human chemokine system is comprised of 50 chemokines and 20 chemokine receptors [6]. They are classified into four different categories, namely, CXC, CC, C and CX3C on the basis of the pattern of cysteine residues in the chemokine ligands [6]. CXCR4 is specific for chemokine ligand 12 (CXCL12), which is also called stromal-derived-factor-1 (SDF-1). As a homeostatic chemokine, SDF-1 is an 8 kDa chemokine peptide with 67 amino acid residues, mainly localized in bone marrow stromal cells. Besides CXCR4, SDF-1 has also been reported to interact with CXCR7 [2]. Like other chemokines, SDF-1 binds to its receptors to promote directional migration of cells to specific locations (chemotaxis) [6-9].
The SDF-1/CXCR4 axis is gaining significant attention mainly due to its important roles in pathological processes, such as HIV entry, tumor metastasis, cell trafficking in autoimmune diseases and inflammatory conditions [10]. Therefore, therapeutic intervention of the SDF-1/CXCR4 interaction is promising for a variety of clinical applications.
CXCR4 structure
The CXCR4 structure consists of 352 amino acid residues comprising an N-terminal domain, seven TM domains, three extra-cellular loops (ECL), three intra-cellular loops (ICL) and a C-terminal domain [11]. Crump et al proposed a two-site theory for the binding of the natural ligand, SDF-1 with CXCR4 [12]. First, the RFFESH loop (site 1) of SDF-1 interacts with the N-terminal domain of CXCR4; then the N-terminal region (site 2) of SDF-1 binds to the receptor groove comprising the TM helices and the extra-cellular loops.
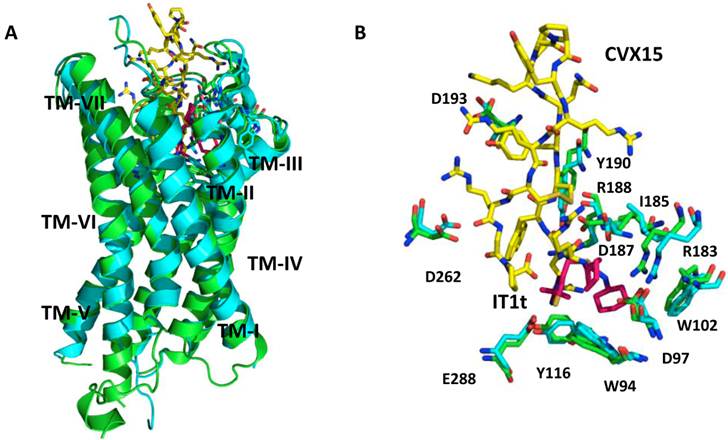
There have been several crystal structures of the CXCR4 protein published. PDB identification codes 3ODU and 3OE0 describe the crystal structures of the TM regions of CXCR4 co-crystallized with a small-molecule inhibitor IT1t and a cyclic peptide inhibitor CVX15, respectively (Figure 2) [11]. Both structures are CXCR4 homodimers, with IT1t or CVX15 located in the ligand-binding cavity that comprises the N-terminal, ECL2, ECL3 and TM domains. It is important to note that the binding cavity of CXCR4 is larger and closer to the extra-cellular surface compared to other GPCRs. IT1t binds in only a portion of the cavity, interacts with TMs I, II, III and VII, while, peptide CVX15 occupies the complete binding cavity and makes contact with all TMs. In addition, CVX15 binding causes conformational changes in the binding cavity, especially at the N-terminus and to some extent the extracellular portions of TMs V, VI and VII [11], whereas IT1t induces no significant conformational changes (Figure 2B).
CXCR4 crystal structures. A. Superimposed CXCR4 PDB structures, 3ODU (green) and 3OE0 (cyan) along with small molecule ligand IT1t (pink) and peptidic ligand CVX15 (yellow); B. Binding site of CXCR4 - a small conformational changes are visible between those two structures in the binding site region. IT1t (pink); CVX15 (yellow); binding site residues from 3ODU (green) and 3OE0 (cyan).
Role of CXCR4 in HIV Infection
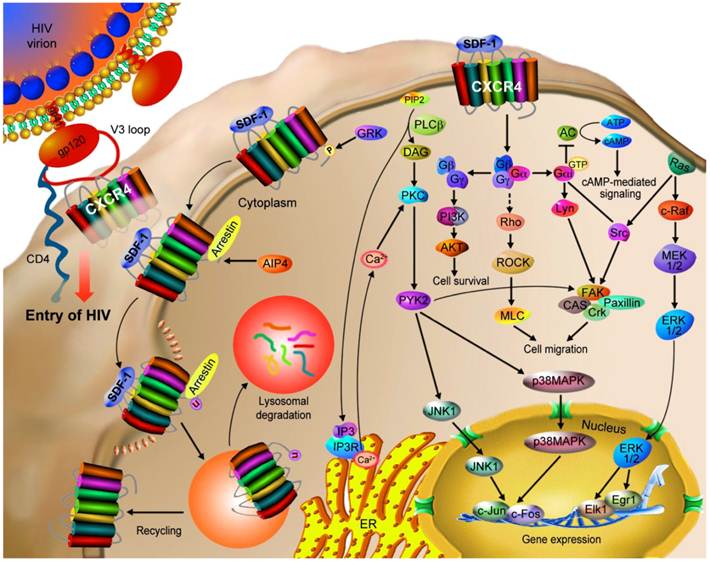
CXCR4 and CCR5 are the two major co-receptors for HIV entry into its target cells in the human immune system and play important physiological roles in viral infection (Figure 3) [13, 14]. In a multi-step process, HIV enters the target cells by binding to the host surface receptor CD4 and a co-receptor, either CCR5 or CXCR4 [13]. As the initiation step, viral glycoprotein gp120 interacts with CD4, which in turn triggers the binding of gp120's V3 loop to the N-terminus, ECL2, ECL3 and the ligand binding cavity of CXCR4 [11]. These interactions lead to a conformational change in the viral TM protein gp41, causing a pH-dependent fusion of the viral and the host cell membranes and the delivery of the viral payload [15-18]. In early stages of HIV infection, HIV predominantly uses the CCR5 co-receptor, whereas during the disease progression HIV uses either CXCR4 alone or in combination with CCR5 in about 50% of the infected individuals [18, 19]. Use of CXCR4 as a co-receptor is associated with a marked drop in CD4+ T-cell counts [19]. Unfortunately, individuals infected by CXCR4 utilizing strains experience a faster rate of disease progression [20, 21].
CXCR4 mediates HIV infection and cancer progression. CXCR4 is a co-receptor used along with CD4 by HIV-1 strains for infecting T cells. The binding of gp120 to CD4 induces a conformational change of gp120, allowing it to interact with CXCR4's N-terminal, ECL2 and ECL3 domains as well as the ligand binding cavity through the V3 loop of gp120. These interactions trigger a conformational change in gp41, causing a pH-dependent fusion of the viral and the host cell membranes and thus the delivery of the viral payload. CXCR4 is also involved in the progression of tumor (hematopoietic and solid) via the interaction with SDF-1. The SDF-1/CXCR4 interaction stimulates tyrosine phosphorylation of CXCR4, followed by the activation of multiple G protein-dependent signaling pathways, which may be different among cell types. The major potential pathways and their cellular outcomes are described in this figure. Activated CXCR4 recruits arrestin 2/3, which mediate receptor internalization. CXCR4 may then be recycled to the cell surface or ubiquitinated by AIP4 that results in the lysosomal degradation of CXCR4.
Role of CXCR4 in Cancer Cell Metastasis
CXCR4 is found to be a prognostic marker in many different cancers [22], including leukemia [23-27], breast [28, 29], lung [30, 31], prostate [32], ovarian [33] and colorectal cancers [34], where the SDF-1/CXCR4 axis plays an important role in cancer progression [22] (Figure 3). In addition, the organs and tissues that possess high levels of SDF-1, such as liver, lung, bone marrow, and lymph nodes, attract the migration of CXCR4-expressing cancer cells [22].
Chronic Lymphocytic Leukemia (CLL): CXCR4 expressing neoplastic B-cells migrate towards bone marrow stromal cells and lymphoid nurse-like cells that secrete high levels of SDF-1, in a CXCR4 dependent fashion [23]. In the bone marrow microenvironment, CLL cells are protected from spontaneous or drug-induced apoptosis, resulting in resistance to conventional chemotherapy. Combination therapy with CXCR4 antagonists, such as plerixafor (Mozobil/AMD3100, Genzyme Corp.) and T140 analogs, disrupts the adhesive interaction between CLL and stromal cells, recirculates CLL cells towards the bloodstream and thus exposes these neoplastic cells to conventional drugs [35]. Plerixafor has been approved by the FDA as the first small-molecule CXCR4 antagonist for use in combination with granulocyte-colony stimulating factor (GCSF) to mobilize hematopoietic stem cells to the bloodstream for collection and subsequent autologous transplantation in patients with non-Hodgkin's lymphoma and multiple myeloma.
Breast Cancer: CXCR4 plays an important role in breast cancer progression by enhancing tumor growth, tumor cell invasion, and metastasis to bone and lung. Inhibition of SDF-1/CXCR4 interaction using an anti-CXCR4 monoclonal antibody significantly impairs the migration of breast cancer cells to regional lymph nodes and lung in nude mice [28]. Moreover, CXCR4 gene silencing using small interfering RNAs (siRNA) inhibits breast cancer metastasis in mouse models [36].
Small Cell Lung Cancer (SCLC): Overexpression of CXCR4 is found in SCLC [30, 31, 37] and mediates the migration of SCLC to bone marrow along an SDF-1 gradient [37]. The expression level of CXCR4 can be used as an independent prognostic factor for disease free survival of SCLC patients [37].
Prostate Cancer: CXCR4 is important for the invasion and metastasis of prostate cancer cells to the bone marrow [32, 38] and it is also a prognostic factor for prostate cancer patients [39]. In a clinical study it was observed that prostate cancer patients with higher levels of CXCR4 had lower survival rates [39].
Ovarian Cancer: CXCR4 is overexpressed in human advanced and metastatic ovarian cancer. Metastasis of epithelial ovarian cancer (EOC) is regulated by the SDF-1/CXCR4 axis. Plerixafor treatment significantly retarded SDF-1 mediated invasion and metastasis of EOC cells [33].
Melanoma: The SDF-1/CXCR4 axis plays a crucial role in the metastasis of melanoma cells [40, 41]. By secreting SDF-1, lymphatic vessels guide the metastasis of CXCR4+/CD133+ melanoma cells towards target organs [40]. Plerixafor in combination with the chemotherapeutic agent dacarbazine significantly suppressed the metastatic phenotype of melanoma compared to dacarbazine alone [40].
CXCR4 and Rheumatoid Arthritis
SDF-1 acts as a pro-inflammatory cytokine through CXCR4 in the development of rheumatoid arthritis (RA) [42-44]. Thymocytes and T-cells from lymph nodes migrate towards SDF-1 in a CXCR4 dependent fashion, implicating CXCR4's role in T-cell chemotaxis [42]. Increased accumulation of T-cells in arthritic joints was observed to be associated with the expression of CXCR4 in T-cells [42, 44]. SDF-1 is expressed in synovial epithelial cells and it has been reported that persistent induction of CXCR4 by stromal derived factors, such as TGF-β led to SDF-1 mediated accumulation of CD4 T cells within the rheumatoid joint [44]. CXCR4 antagonist, plerixafor was effective in reducing collagen-induced arthritis in a mouse model [43]. Moreover, there is also a reduction in collagen-induced arthritis in CXCR4 deficient mice [42].
CXCR4 and WHIM Syndrome
WHIM (Warts, Hypogammaglobulinemia, Infections, and Myelokathexis) syndrome is a rare congenital hematological immunodeficiency disorder in which mature neutrophils fail to reach blood circulation from bone marrow. Dysfunction of the SDF-1/CXCR4 signaling axis is a major cause of WHIM syndrome because it hinders the development of B-lymphocytes and myeloid cells [45]. Inherited mutations in the gene encoding CXCR4 have been also reported to correlate with the disorder [45, 46].
CXCR4 and Ischemic Injury
Following coronary artery occlusion, SDF-1α level is increased in the infarcted area, resulting in the recruitment of progenitor cells expressing CXCR4 for post-infarct repair [47]. GCSF and hypoxia inducible factor-1α (HIF-1α) have been shown to up-regulate SDF-1α in the infarcted area in a rabbit ischemia model and promote the migration of CXCR4 expressing cells towards cardiac tissues [47-49]. Plerixafor is able to inhibit this protective effect [47, 49].
CXCR4 and Pulmonary Fibrosis
The SDF-1/CXCR4 axis plays a pivotal role in mobilizing bone marrow derived stem cells and inflammatory cells. Recruitment of circulatory fibrocytes and bone marrow derived progenitor cells during lung injury is guided by the SDF-1/CXCR4 axis promoting the pathogenesis of pulmonary fibrosis. Treatment with plerixafor prevents pulmonary fibrosis in bleomycin-induced murine pulmonary fibrosis. Increased expression of CXCR4 was observed in the lungs of bleomycin-treated mice, whereas treatment with plerixafor significantly decreased SDF-1 in bronchoalveolar lavage fluids at an early stage and CXCR4 expression at the later stage after bleomycin injury [50].
CXCR4 and Systemic Lupus Erythematosus
Overexpression of CXCR4 along with SDF-1 promotes the pathogenesis of systemic lupus erythematosus (SLE) [51-53]. CXCR4 is significantly up-regulated in monocytes, neutrophils, and B-cells from murine models of lupus [53] as well as patients with SLE [51]. SDF-1 is predominantly overexpressed in skin and kidney, making them the final destinations of CXCR4-expressing cells. Treatment with CXCR4 antagonists significantly improved disease conditions and nephritis in SLE mouse models [52].
Discovery of CXCR4 Antagonists
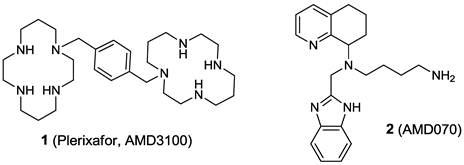
The important roles of CXCR4 in multiple diseases have encouraged the development of clinically viable CXCR4 antagonists. This has resulted in the FDA approval of the first CXCR4 antagonist, plerixafor (1) for the mobilization of hematopoietic stem cells (Figure 4) [54]. TG-0054 (structure not disclosed publicly) is an injectable small molecule CXCR4 antagonist that is currently in phase I/II clinical trials for multiple myeloma, non-Hodgkin lymphoma, and Hodgkin disease. Another small molecule, AMD070 (2), an orally active CXCR4 antagonist, is under clinical investigation for the prevention of T-tropic HIV infection (Figure 4). Newer CXCR4 antagonists are under clinical evaluation for therapeutic intervention in various cancers, hematologic and vascular disorders, HIV infection, and other immune system disorders involving the modulation of the SDF-1/CXCR4 axis, such as rheumatoid arthritis and lupus [54-56].
The identification and development of CXCR4 antagonists have been described in several recent studies [57-61]. In 2008, we published a comprehensive review covering publications and patent applications describing small molecules endowed with CXCR4 antagonistic activity [17]. The earliest efforts to find CXCR4 antagonists focused mainly on peptide derivatives. Despite the disadvantages in terms of pharmacokinetic properties, these peptides have been instrumental in defining a basic pharmacophore model for designing smaller molecules. Subsequently, numerous compounds have been shown to significantly inhibit CXCR4. In general, these compounds are cationic molecules able to bind the predominantly anionic extracellular domain of CXCR4. They belong to different chemical classes including cyclic penta- and tetra-peptides, diketopiperazine mimetics, bicyclams, tetrahydroquinolines, thiazolylisothiourea derivatives, benzodiazepines, dipicolylamine-zinc(II) complexes and naturally occurring derivatives.
Herein, we focus on small molecules claimed to be CXCR4 antagonists described in the literature between 2008 and 2012. Several previously identified lead molecules were modified over this period to enhance their therapeutic properties. To date, although more than 20 different chemical classes have been claimed as CXCR4 antagonists, only a few have been described in detail including tetrahydroquinolines, N-substituted indoles, 1,4-phenylenebis(methylene) derivatives, and N-containing heterocycles.
Cyclic Pentapeptide-based Antagonists
Several earlier studies conducted over the last ten years have led to the identification of a highly potent CXCR4 antagonist, a β-sheet-like 14-mer peptide with a single disulfide bridge, known as T140. Intensive structure-activity relationship (SAR) studies on this peptide derivative allowed the determination of four amino acid residues indispensable for activity: Arg2, L-3-(2-naphthyl)alanine3 (Nal3), Tyr5 and Arg14. These key residues are spatially close in the T140 three-dimensional structure and are particularly positioned across the disulfide bridge. The subsequent pharmacophore approaches used for further molecular-size reduction led to the identification of cyclic pentapeptides containing these indispensable amino acids.
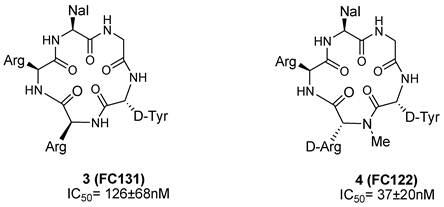
Using both conformation-based and sequence-based libraries, a potent CXCR4 antagonist, FC131, (3) was identified with anti-HIV activity [62-65]. SAR studies performed on FC131 analogues, such as modification of the amino acid residues as well as ring structures and backbones, indicated that a defined spatial arrangement of the functional groups on the side chain is necessary for the activity of such compounds. For example, N-methylation of the peptide bonds of FC131 and the configuration of epimeric derivative significantly affected its activity [66-68].
In an attempt to identify optimized compounds, an appropriate combination of sequence, chirality, and introduction of suitable functional groups were explored. Systematic SAR studies, focusing on sequence optimizations, were conducted to identify essential structural and electrostatic requirements for the activity of FC131. For example, the replacement of an Arg residue with the epimeric N-methyl-D-arginine led to the identification of the cyclic pentapeptide-based CXCR4 antagonist FC122 (4) that shows a significant enhancement of CXCR4 antagonistic activity (Figure 5) [11, 66, 69, 70].
Structures of select cyclic pentapeptide-based CXCR4 antagonists. IC50 values are the concentrations for 50% inhibition of the [125I]-SDF- 1α binding to CXCR4 transfectant of HEK293 cells [70].
It is now well known that a systematic replacement of the planar peptide bond with an alkene bioisostere is useful for the estimation of the electrostatic effects and for the SAR evaluation of peptide derivatives. In particular, due to their electrostatic and steric properties, the fluorinated or substituted alkene isosteres represent more appropriate peptide bond mimetics than the unsubstituted alkene isosteres [71-76].
On the basis of these considerations, the effects of the Arg2-Arg3 and Arg3-Nal4 peptide bonds on the activity of FC131 have been investigated through the synthesis and evaluation of the alkene analogues such as compounds 5-8 (Figure 6) [69].
Structures of representative examples of cyclic pentapeptide-based CXCR4 antagonists [69].
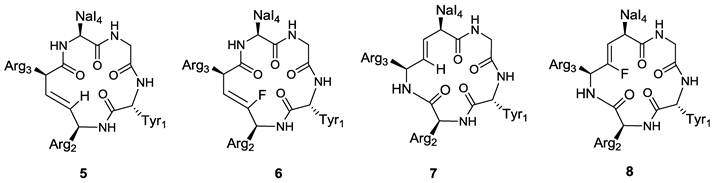
In order to evaluate the electrostatic effects of the amide carbonyl groups within the peptide bonds on the activity of FC131, a comparative study using unsubstituted and fluorinated alkene isosteres has been attempted. Both (E)-alkene and (Z)-fluoroalkene analogues (5 and 6) with substitutions at the Arg2-Arg3 dipeptide moderately inhibited SDF-1 binding to CXCR4 with IC50 values of 1.46 and 1.78 μM, respectively. Their potencies were approximately 20-fold lower than the parent compound (IC50 (FC131) = 0.068 μM), indicating an important role of the amide bond within the Arg2-Arg3 dipeptide and suggesting that the lower activity may be attributed to the loss of the H-bonding.
The replacement of the Arg3-Nal4 peptide bond with an (E)-alkene isostere in compound 7 produced a negative effect on CXCR4 inhibitory activity, probably due to the lack of H-bonding of amide moiety and a different interaction of pseudo-1,3-allylic strain between the Arg3 carbonyl group and the Nal4 side chain. The substitution of the Arg3-Nal4 dipeptide with the (Z)-fluoroalkene moiety in compound 8 also led to the loss of CXCR4-binding activity (IC50 (7,8) > 10 μM). The amide hydrogen within the Arg3-Nal4 of FC131 was shown to be necessary for the ligand-CXCR4 interaction, giving important suggestions for the design and the development of novel cyclic pentapeptide based CXCR4 antagonists.
In a separate study, the substitution of peptide bond carbonyl (CO) groups with imino (C=NH) groups has been taken into consideration and a series of amidine-containing FC131 analogues (9-15), in which each peptide bond was replaced with the amidine substructure, have been designed (Table 1) [70, 77]. Peptides 9-13 displayed higher activities than both FC131 and FC122 (cf, Table 1 and Figure 5), indicating that the basic amidine moiety possesses the expected requirement to increase the affinity for the receptor. On the other hand, substitution of the Tyr-Arg dipeptide reduced the CXCR4 antagonistic property in compounds 14 and 15. These observations demonstrate that the D-Tyr-Arg peptide bond is crucial for the peptide conformation and thus for an optimized interaction with the targeted receptor. Nal-Gly modification resulted in the most potent inhibitors (10 and 11) of this series of compounds, suggesting that the amidine substructure is more appropriate than the compact amide scaffold. Both stereoisomers of Nal-Gly- and Tyr-Arg- modified analogues (10, 11 and 14, 15) showed similar antagonistic activities (IC50(10) = 4.2 nM compared to IC50(11) = 4.9 nM; IC50(14) = 679 nM compared to IC50(15) = 334 nM).
Inhibitory activity of FC131 derivatives against SDF-1α binding to CXCR4.
| Compd | a.a sequence | IC50 (nM) |
|---|---|---|
| 9 | Cyclo(-D-Tyr-Arg-Arg-Nal-Gly-Ψ-) | 9.4±3.0 |
| 10 | Cyclo(-D-Tyr-Arg-Arg-Nal-Ψ-Gly-) | 4.2±0.31 |
| 11 | Cyclo(-D-Tyr-Arg-Arg-D-Nal-Ψ-Gly-) | 4.9±1.1 |
| 12 | Cyclo(-D-Tyr-Arg-Arg-Ψ-Nal-Gly-) | 11±2.9 |
| 13 | Cyclo(-D-Tyr-Arg-Ψ-Arg-Nal-Gly-) | 16±7.2 |
| 14 | Cyclo(-D-Tyr-Ψ-Arg-Arg-Nal-Gly-) | 679±132 |
| 15 | Cyclo(-Tyr-Ψ-Arg-Arg-Nal-Gly-) | 334±6.2 |
| 16 | Cyclo(D-Phe(4-F)-Arg-Arg-Nal-Gly-) | 220 |
| 17 | Cyclo(D-Phe(4-F)-D-Arg-Arg-Nal-Gly-) | 310 |
| 18 | Cyclo(Phe(4-F)-Arg-Arg-Nal-Gly-) | 220 |
| 19 | Cyclo(Phe(4-F)-D-Arg-Arg-Nal-Gly-) | 2200 |
| 20 | Cyclo(D-Phe(4-Cl)-Arg-Arg-Nal-Gly-) | 1200 |
| 21 | Cyclo(D-Phe(4-Br)-Arg-Arg-Nal-Gly-) | 2300 |
| 22 | Cyclo(D-Phe(2-F)-Arg-Arg-Nal-Gly-) | 59 |
| 23 | Cyclo(D-Phe(3-F)-Arg-Arg-Nal-Gly-) | 88 |
| 24 | Cyclo(L/D-Phenylglycine-Arg-Arg-Nal-Gly-) | 1100 |
| 25 | Cyclo(β-(2-thienyl)-D-Ala-Arg-Arg-Nal-Gly-) | 56 |
| 26 | Cyclo(Arg-D-Phe(4-F)-Arg-Nal-Gly-) | 35 |
| 27 | Cyclo(Arg-D-Phe(4-Cl)-Arg-Nal-Gly-) | 790 |
| 28 | Cyclo(Arg-D-Phe(4-Br)-Arg-Nal | 570 |
| 29 | Cyclo(Arg-D-Phe(4-NO2)-Arg-Nal-Gly-) | 940 |
| 30 | Cyclo(D-Tyr-His-Arg-Nal-Gly-) | 37 |
| 31 | Cyclo(D-Tyr-D-His-Arg-Nal-Gly-) | 35 |
IC50 values are the concentrations for 50% inhibition of the [125I]-SDF- 1α binding to CXCR4 transfectants of HEK293 cells (compounds 9-15) or CHO cells (compounds 16-31). Ψ indicates the [[-C(=NH)-NH-]] substructure; Nal indicates 3-(2-naphthyl)alanine.
These results suggest an improved activity of amidine containing compounds due to a higher flexibility when compared with former cyclic pentapeptides. This study not only establishes that the substitutions of the peptide bonds in FC131 (except for the D-Tyr-Arg position) with an amidine motif improves the CXCR4 inhibitory activity, but also demonstrates that these analogues are selective antagonists for CXCR4 without affecting CXCR7 or CCR5 activity.
A parallel study was performed focusing on the exploration of amino acid residues of cyclic pentapeptides by systematic substitutions of various aromatic amino acid residues [62]. In order to explore new pharmacophore moieties, FC131 was used as a lead compound for the development of novel CXCR4 antagonists. Ever since earlier studies showed for compound 16, where D-Tyr1 was replaced by 4-fluoro-D-phenylalanine, a certain CXCR4-binding activity, three more 4-fluorobenzoyl derivatives, compounds 17-19, were synthesized to evaluate the roles of amino acids at positions 1 and 2 [62]. Although the CXCR4 antagonistic activities of these analogues are lower compared to FC131, all of them, except for compound 19, showed high binding activity (IC50 range: 0.2-0.3 μM). On the other hand, compound 19 showed only a moderate activity (IC50 = 2.2 μM), suggesting that the combination of Phe(4-F)1 and D-Arg2 was not optimal. Starting from compound 16, several derivatives, in which the D-Phe(4-F)1 moiety was replaced by various aromatic amino acids, while maintaining the same configuration of [D-Phe(4-F)1, L-Arg2] fragment, have been synthesized. The order of preference among halogen atoms as a substituent at position 4 of D-Phe1 was shown to be F then Cl then Br, resulting in the following order of activity: 16 > 20 > 21 (IC50 = 0.22, 1.2 and 2.3 μM, for the three halogen-containing derivatives, respectively) (Table 1). This indicates that a small or electron-withdrawing group at position 4 on D-Phe1 positively affects the activity of this class of molecules. Subsequently, the order of preferred positions of fluorine on the phenyl ring of D-Phe1 was determined to be as ortho, meta, para (22 > 23 > 16; IC50 = 0.059, 0.088 and 0.22 μM, respectively). In addition, the thienyl group is suitable for the side-chain of the amino acid at position 1, as is evident from the high CXCR4 binding activity of compound 25, [β-(2-thienyl)-D-alanine(D-Thi)1]-2, but not compound 24. A further series of analogues with the sequence of Arg1-aromatic D-amino acid in position 2 were prepared. Among the halogen substituents at position 4 of the phenyl ring within D-Phe2, fluorine showed to be optimal, since chlorine and bromine, or even a nitro group, negatively affected the activity profile of 26, 27, 28 and 29 (IC50 = 0.035, 0.79, 0.57 and 0.94 μM, respectively). Furthermore, in order to improve both basic and aromatic characters of such derivatives, two analogues, in which the Arg2 of FC131 was replaced by an L/D-His residue, compounds 30 and 31 were prepared and turned out to be potent CXCR4 antagonists with IC50 values of 0.037 and 0.035 μM, respectively. All the data reported in this study indicate that introductions of aromatic and basic substituents on amino acid side chains are useful to determine the antagonists/CXCR4 binding mode on the cell membrane.
Structures composed of dimers and multimers have been extensively used in chemistry [78, 79], especially in molecular imaging [80]. Previously, dimeric analogues of FC131 were used as starting points to evaluate the optimal distance between the monomeric units [81]. In early studies the CXCR4 was assumed to contain two neighboring binding sites, respectively responsible for the affinity and the signaling function. Thus, the identification of dimeric ligands would be a promising approach for the development of effective CXCR4 antagonists [12]. Recently, the crystal structure of CXCR4 complexed with small-molecule antagonists was reported [11, 82]. This offered the opportunity to model the interaction of novel derivatives with the receptor. An improved binding affinity was detected after the presence of long spacers, with lengths from 20 to 80Å, in a dimeric ligand suggesting two different binding sites [11, 82].
After further modifications of FC131, including an N-methylation and the introduction of a side chain, aimed at affinity enhancement, compound 32 was selected for a dimerization study. The side chain of D-[NMe]Orn2 represents, within the peptide, the “least important residue”, amenable to acylation, and therefore it has been used as an anchor point for dimerization, leading to derivative 33 (Figure 7) [81, 83].
Structures of representative examples of a monomeric cyclic pentapeptide-based CXCR4 antagonist, two dimeric derivatives and a dimeric Ga-labeled DOTA complex [81].
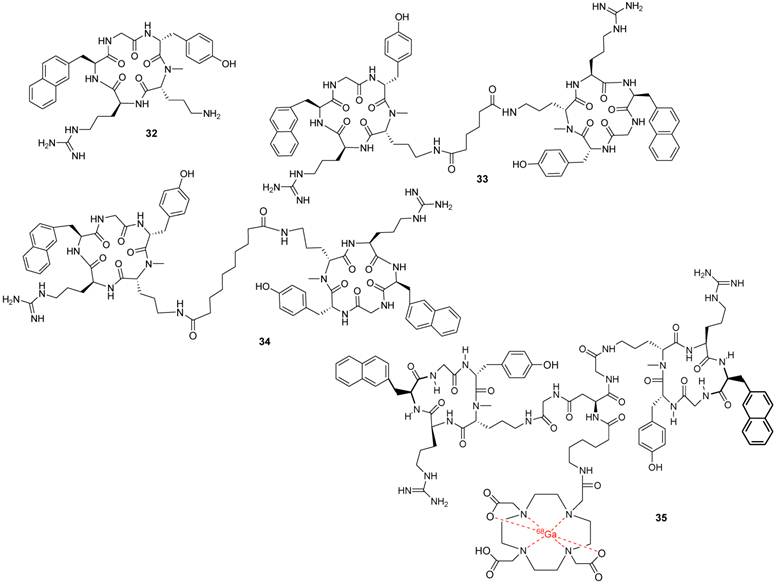
Nine dimers were synthesized and used to elucidate the optimal spacer length as well as the binding mode. The best results were achieved with a 5-10 carbon-atom spacer. Compound 34 was identified as the best dimer derivative with an IC50 value of 2 nM that was 3-fold more potent than the monomer 32, from which it was synthesized. A possible explanation for the higher binding affinity could be the presence of a sub-binding site for the second peptide unit close to the main binding site. This was also confirmed by the structural elucidation of the pharmacophoric groups in the dimers and with a comparison of the dimeric scaffold to the oligomer polyphemusin II structure that was used for the design of the first cyclic-pentapeptide derivatives and by recent molecular modeling studies using the CXCR4 crystal structure [11].
In the further elaboration, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) was introduced as a chelator for molecular imaging studies. The DOTA conjugates and some of their corresponding metal complexes retained good CXCR4 affinity, and one 68Ga-labeled compound (35) was further studied as a PET tracer. Organ analyses revealed high accumulation of the labeled compound in the liver, probably due to its high lipophilic character [81].
Indole-based CXCR4 Antagonists
A recent study focused on the design of novel non-peptide CXCR4 antagonists based on the SAR and conformational studies of cyclic pentapeptides (Figure 8) [84]. The positioning of key functional groups and the conformational analysis were performed using a template constructed from the rigid backbone of these latter compounds. Previous SAR studies on pentapeptides showed that at least three functional groups on the peptide side-chains are required: an aromatic ring (such as 2-naphthyl- or 3-indolyl group), one or two guanidino groups, and a phenol group. Due to the free rotation of the side-chains, it was not straightforward to define the mutual spatial relationships among the functional groups, although previous studies had reported the fixed distances between Cβ atoms substituted with key functional groups on the rigid cyclic peptide nucleus. [65, 66].
Structures of indole-based CXCR4 antagonists [84].
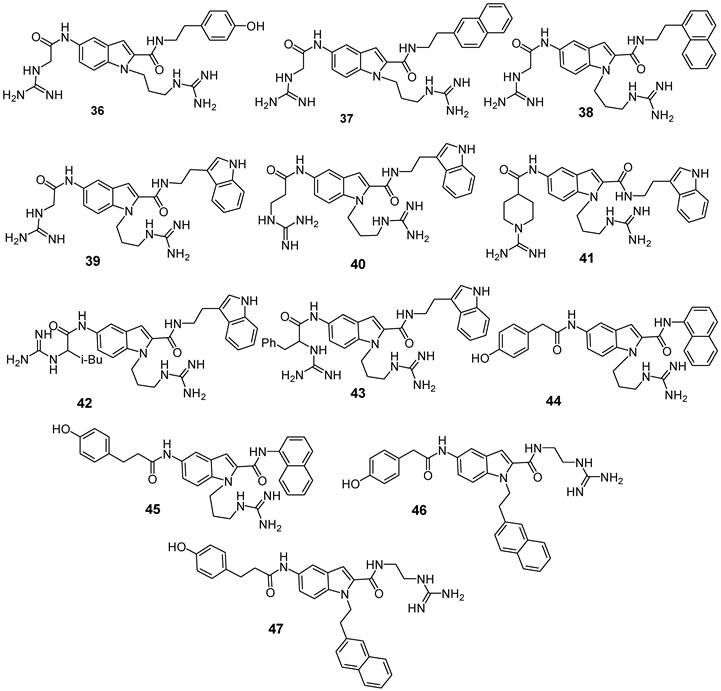
In order to identify novel non-peptide CXCR4 ligands, the introduction of crucial functional moieties for receptor binding onto a bicyclic heterocyclic scaffold to mimic FC131's quite rigid backbone was carried out. Among several molecular scaffold candidates, 5-aminoindole-2-carboxylic acid was selected since molecular modeling experiments showed that it meets the spatial requirements for the three key substituents. Moreover, indoles represent an important class of bioactive compounds whose properties are well documented. Various synthetic approaches for the introduction of substituents on these indoles are available. Among the 12 indole derivatives synthesized (compounds 36-47, Figure 8), compound 39, with 2-(3-indolyl) ethyl group at the R2 position, showed the highest potency with an IC50 value of 3.0 μM and 88% of inhibition at 10 μM compared to analogues having a (4-hydroxyphenyl)-, (1-naphthyl)- or (2-naphthyl)-alkyl group at the R2 position (compounds 36-38: inhibition at 10 μM = 23-63%). Chain elongation of the guanidinoacetyl group (R3) of 39 induced a modest reduction of binding affinity (inhibition of compound 40 = 70% at 10 μM). The introduction of a N-amidinopiperidine-4-carbonyl substituent was also acceptable for a higher potency (IC50(41) = 3.0 μM). Introduction of an isobutyl or benzyl group at the α-carbon within the guanidine-methyl carbonyl group of compound 39 did not cause significant decrease of the binding affinity (compounds 42 and 43). When derivatives were resolved in single enantiomers, compounds with S-configuration at the chiral center showed higher CXCR4 antagonistic activity with respect to the corresponding R-isomers. (S)-42 was identified as the most potent compound [IC50((S)-42) = 1.2 μM]. As a further step, a comparison of energy-minimized structures of (S)-42 and previously reported conformation of FC131 was carried out. As expected, such studies confirmed that three functional groups on the indole moiety properly overlapped the three pharmacophore residues of the parent compound. Accordingly, indole scaffold closely resembles the Arg-Arg-Nal sequence on backbone of FC131. Taken together, these data suggest that indole derivatives, although their IC50 values are in the micromolar range, represent functional leads for further development of novel CXCR4 antagonists useful in medicinal chemistry programs.
AMD070 Derivatives: Tetrahydroquinolines-based CXCR4 Antagonists
Bicyclams were the first class of non-peptidic small molecules targeting CXCR4. They were recognized as potent anti-HIV compounds before their mechanism of action was completely established. Although these derivatives show significant potency, their clinical application is limited by poor oral bioavailability [85]. This might be due to both the large size and the cationic nature of these compounds. Subsequently, derivatives with pharmacokinetic properties better than the original compounds were synthesized. To improve the pharmaceutical properties of bicyclams, one or both rings were replaced by smaller heterocyclic groups.
Several studies have dealt with the development of tetrahydroquinoline-based derivatives. The unique structural feature of these compounds is the presence of a core structure, a substituted (R), (S) or (RS) (N`-(1H-benzimidazol-2-ylmethyl)-N`-5,6,7,8-tetrahydroquinolin-8-yl-1,4-alkylamine) (48) that replaces the macrocyclic nucleus of bicyclams. Among this class of derivatives, compound 2 (AMD070, AnorMED) is particularly notable due to its significant anti-HIV efficacy in human clinical studies [86]. Key features of the AMD070 pharmacophore include a triad of basic nitrogen atoms and a distal amino group linked to the central nitrogen via a 4-carbon tether. Several studies were directed towards the improvement of the antiviral potency and/or the ADME properties of compound 2 through structural modifications [87-93]. Some studies explored the possibility to shift the distal amine side chain from the central nitrogen to the benzimidazole ring with retention of antiviral activity. In particular, a series of N-(1H-benzimidazol-2yl-methyl)-5,6,7,8-tetrahydro-8-quinolinamine derivatives, with basic side chains attached to the N-1 and C-4 position of the benzimidazole, were synthesized. The optimal spacer length between the benzimidazole N-1 and the basic nitrogen is a 3-carbon propylene moiety (e.g. 49), while shorter (2 carbon) or longer (4-5 carbon) linkers result in 5 to 10-fold lower anti-HIV activity. The tertiary amine 52 and the guanidine derivative 53 showed an acceptable anti-HIV activity, while the trifluoroacetyl derivative 54 exhibited only a weak activity. The structures of these tetrahydroquinoline derivatives are shown in Figure 9 [93].
General structure (48) and representative examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values were determined in antiviral assay using HOS cells expressing hCXCR4/hCCR5/hCD4/pHIV-LTR-luciferase and HIV-1 IIIB strain [93].
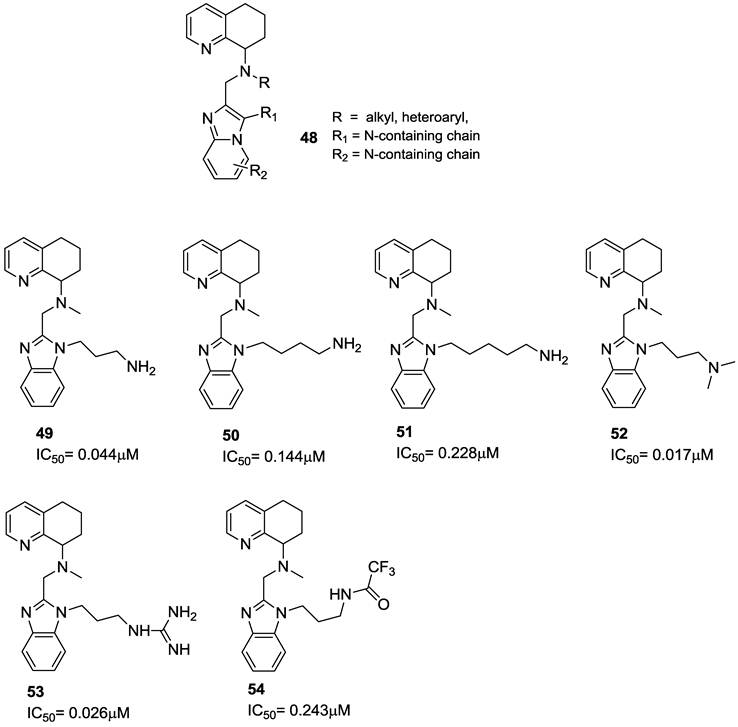
The effect of shifting the basic side chain from the N-1 nitrogen to the C-4 position of the benzimidazole was investigated. For these amide-linked analogues, the ethylene diamine derivative 55 showed the best activity; in comparison, amides 56 and 57 were about 5 to 10-fold less potent. While 61 and 62 showed only limited activities, compounds 63 and 64 were active in the sub-micromolar range. The introduction of a conformationally constrained piperazine in compound 64 caused a further increase in potency. Remarkable anti-HIV activities were observed with the C-4 linked propylamine derivatives 65-68 as well as the compounds in which the basic amine was incorporated into a heterocycle, such as piperazine (compounds 69-71). In particular, the methylpiperazine derivative 71 showed anti-HIV activity at 2 nM, about 10-fold more active than AMD070. The remarkable anti-HIV activity of the racemic mixture necessitated the synthesis of single enantiomers to identify the active isomer. As a result, the S-enantiomer showed to be very active, while the R-enantiomer was markedly less active. Furthermore, pharmacokinetic studies on this class of derivatives showed that an improved bioavailability was promising to further foster them for clinical use. The structures of these derivatives are shown in Figure 10 [93].
Representative examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values were determined in antiviral assay using HOS cells expressing hCXCR4/hCCR5/hCD4/pHIV-LTR-luciferase and HIV-1 IIIB strain [93].
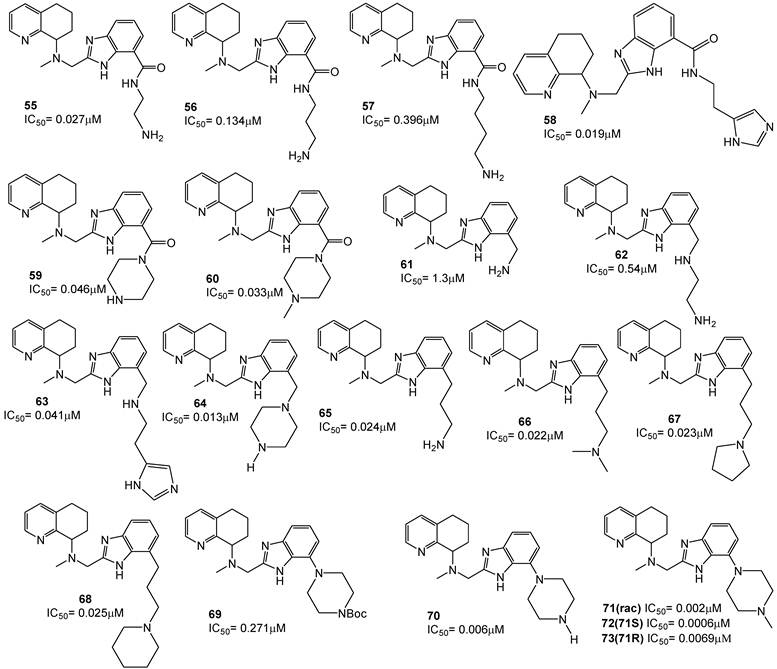
Some other studies have focused on the possibility of replacing the benzimidazole with alternative heteroaromatic ring systems capable of maintaining the required basic nitrogen triad. Accordingly, some benzimidazoles and imidazopyridines have been prepared, and in each series the heterocycles based core was substituted with an amine side-chain, such as a 3-dimethylaminopropyl group (for example compound 74 for the benzimidazole series and 75 for the N-methylpiperazines) [92, 93].
A tricyclic octahydrophenanthroline ring system was in turn incorporated to conformationally restrain the rotation of the 8-amino group within the bicyclic tetrahydroquinoline core. The octahydrophenanthroline ring system includes four distinct stereoisomers. Thus, achiral synthetic methods were first applied in order to obtain all four possible stereoisomers, while a subsequent resolution, via chiral chromatography, has led to the optically active single derivatives (compounds 76 a-d). Testing for anti-HIV activity showed that the cis analogue 76a (anti-HIV IC50 = 35 nM), comparable with bicyclic tetrahydroquinoline derivative 75, was the most potent among the isomers, while the activities of the cis isomer 76b and both trans diastereomers 76c and 76d were more than 10-fold lower.
Subsequently, an imidazopyridine moiety was incorporated (compounds 77a-d) and the SAR trend for these diastereomers was consistent with that observed for the corresponding benzimidazole diastereomers. The imidazopyridine analogue 77a, equipotent with 75, were 50-fold more potent than 77b and about 25-fold more potent than the single trans diastereomer 77c. The structures of these derivatives are depicted in Figure 11 [91].
Select examples of tetrahyidroquinoline-based CXCR4 antagonists. IC50 values were determined in antiviral assay using HOS cells expressing hCXCR4/hCCR5/hCD4/pHIV-LTR-luciferase and HIV-1 IIIB strain [91].
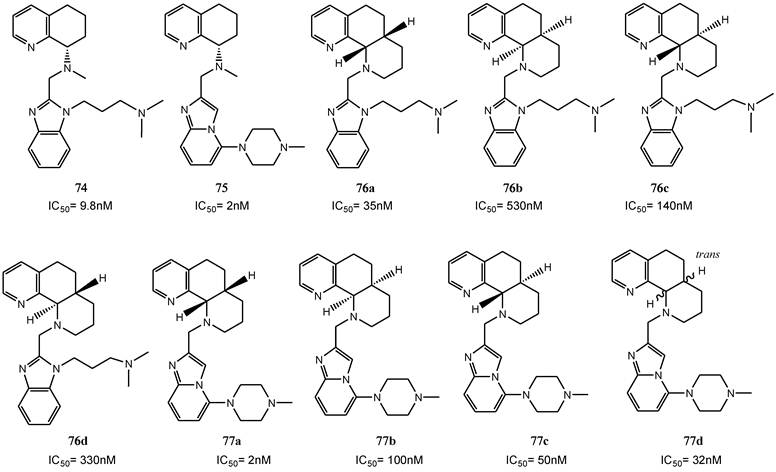
A separate study describes the synthesis and the biological evaluation of derivatives obtained from further modification of AMD070. In particular, the effect of incorporating the distal nitrogen into a piperidine ring, directly attached to the benzimidazole nitrogen, was explored [90]. Ten analogues were prepared (compounds 78-87). The evaluation of the CXCR4 inhibitory activities revealed that cyclization of the amine side chain only moderately affected their antiviral activity. Comparative studies on methylene tethered N-H derivatives 78-80 showed that the five-membered ring is favored for increased activity. The ethylene linked piperidine analogue 84, and the equipotent N-piperidinyl analogues 86 and 87 showed only weak activity. The six-membered ring N-methyl analogue 81 was the most promising compound with an IC50 value of 19 nM. Since two stereocenters were present in the structure, the initially mixture of four stereoisomers was subsequently separated into the single components to evaluate the effects associated with the absolute configuration of the side chains. As a result, the S,S-diasteroisomer showed a seven-fold increase in the antiviral activity with an IC50 of 6 nM. Further modifications focused on the replacement of the side chain methyl group with several alternative substituents. Replacement of the side chain with an isopropyl group led to the most potent compound 88, showing an IC50 of 2 nM for antiviral activity and suitable pharmacokinetic properties.
All these SAR studies on the tetrahydroquinoline series have provided important structure-activity information for the development of suitable CXCR4 antagonists. The structures of these derivatives are depicted in Figure 12 [90].
Select examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values were determined in antiviral assay using HOS cells expressing hCXCR4/hCCR5/hCD4/pHIV-LTR-luciferase and HIV-1 IIIB strain [90].
A more recent study described further modification of AMD070. In particular, the effects of sequentially replacing the benzimidazole ring with a substituted pyridine ring, the opening of the tetrahydroquinoline ring, and the combination of both alterations were investigated to optimize potency. The best result was achieved by attaching a substituted pyridine ring to position 2, according to the previously proposed pharmacophore model [87, 94]. Among the 14 synthesized compounds (89-102), the unsubstituted pyridine analogue 89 was three-fold less potent than the reference compound with an IC50 value of 15.3 nM (versus 5 nM of AMD070), while methyl pyridine derivatives were more active. For example, compound 93, the most potent derivative of the methyl series, showed two-fold higher activity compared to AMD070, with an IC50 value of 2.9 nM, while 92 retained comparable potency (IC50 = 5.3 nM). Interestingly the 3,5-dimethyl analogue 95 was eight-fold more potent than the 5-methyl analogue 92 (IC50 = 0.63 nM). A substitution at the 6-position of compound 93 with either an H-bond donor (amino, 96) or H-bond acceptor (methoxymethyl, 97) moiety resulted in a significant reduction of potency, while substitution at the 3-position was well tolerated (90, 97-102). Other substituents did not significantly affect the potency of these derivatives (103, 104). A further series of analogues (105-114), showing both these changes, were synthesized. Among them, compound 114 displayed an IC50 value of 0.3 nM.
A parallel study reported on the possibility of replacing the benzimidazole with other heteroaromatic ring systems. In order to identify feasible alternative ring systems, various heteroaryl scaffolds with the distal amine side chain attached to the central nitrogen of AMD070 was explored [89]. Most of the reported ring systems led to compounds endowed with noticeable antiviral activity. The isoquinoline derivatives 115 and 116 were particularly promising showing IC50 values of 12 and 11 nM, respectively. Subsequently, the isoquinoline scaffold of 115 was chosen for further study. As a result, the unsubstituted analogue 117 showed significant activity even lacking the amine side chain. The introduction of a 1-carbon primary amine moiety (compound 118) resulted in a significant increase in activity compared to compound 117. On the other hand, the dimethylation of the amino group led to a loss in activity (compound 119). The guanidine derivatives 120 and 128 showed remarkable activities and the best cytotoxicity profiles within the whole series. An interesting aspect of the SAR in this series of compounds was the unexpected high activity of the non-basic derivatives 121, 129 and 130. The dimethylaminoglycine analogue 122 was relatively potent with an IC50 of 12 nM. Proline derivatives 123 and 124 showed a consistent reduction in activity in both the enantiomeric forms. The 3-carbon amine derivative 127, with an IC50 of 5 nM, was the most active analogue among this series of compounds. The structures of derivatives 89-130 are shown in Figures 13-15.
Representative examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values are the concentrations required to protect 50% of MT4 cells against X4 HIV-1 NL4.3 cytopathicity [87].
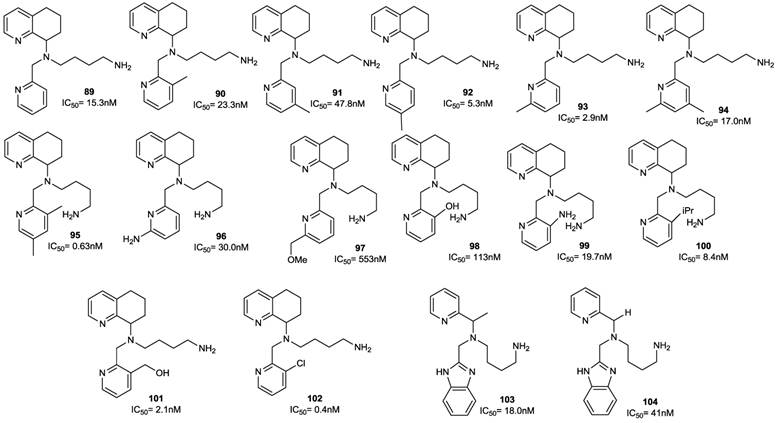
Select examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values are the concentrations required to protect 50% of MT4 cells (compounds 105-114) or HOS cells (compounds 115, 116) against viral cytopathicity [87, 89].
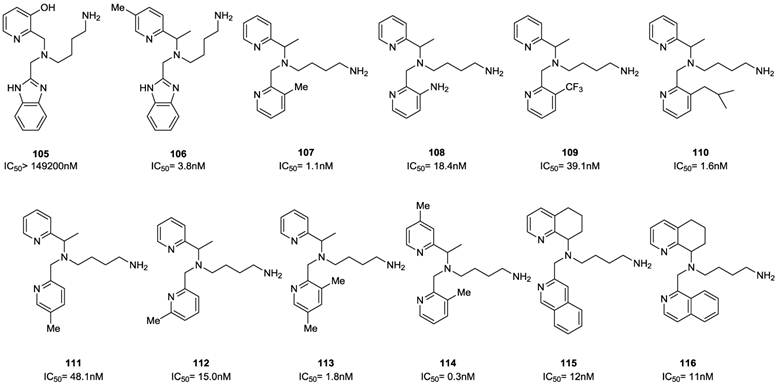
Select examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values were determined in antiviral assay using HOS cells expressing hCXCR4/hCCR5/hCD4/pHIV-LTR-luciferase and HIV-1 IIIB strain [89].
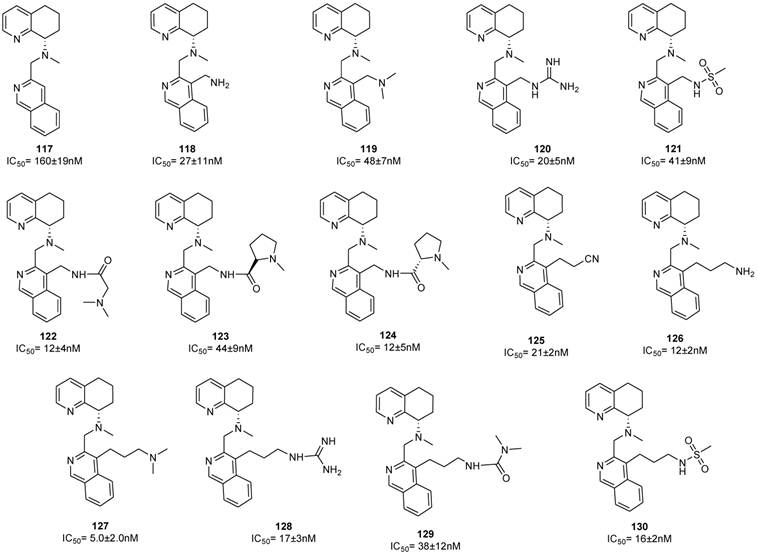
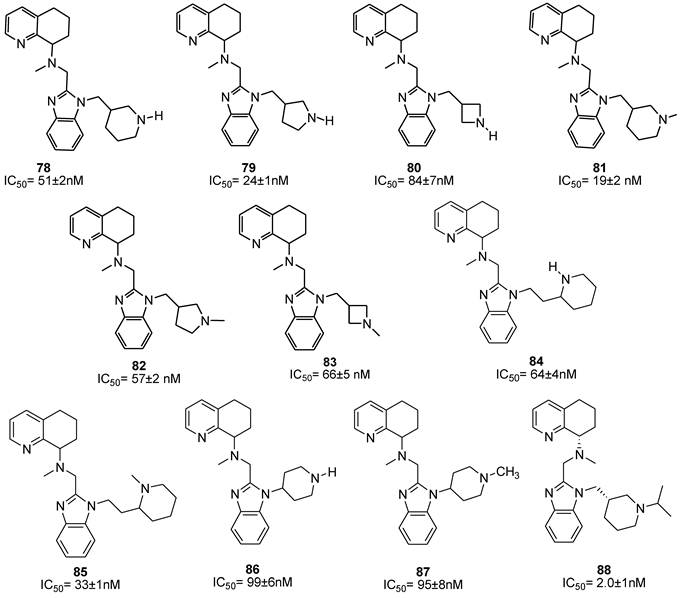
CXCR4 antagonists in which a substituted benzylic aminomethyl group or a heterocyclic aminomethyl moiety replacing the butyl amine residue showed potent antiviral activity [88]. Although these molecules contain a chiral center, the SAR studies were carried out on the racemic mixtures. The introduction of an aminomethyl moiety led to the active compound 135 with an IC50 value of 2 nM. However, the double positive charge at the physiological pH precludes a complete absorption, as showed by preliminary pharmacokinetic studies. In an attempt to reduce this charge, the O-methyloxime derivative 142 was prepared, but this resulted in loss of activity. On the other hand, in order to reduce the basicity of the amine, addition of electron withdrawing groups on the phenyl ring, such as F (e.g. 132) and CF3 (e.g. 133), led to compounds with high absorption potentials, but, with a concomitant loss of antiviral activity.
A comparable potency was achieved by incorporation of a hydroxymethyl or methoxymethyl moiety (compounds 136 and 137), even though the permeability was not improved. Thus, the primary amide 140 resulted in an improvement in potency whereas the acid 138 and methyl ester 139 were less potent. Unfortunately, no satisfactory results were achieved for increasing potency without loss of permeability. Successive structural modifications were focused on heterocyclic replacements of the phenyl group. The introduction of a 2,3-disubstituted pyridine ring with an aminomethyl moiety at the 2-position led to compound 142 with an IC50 value of 0.021 μM that showed a comparable potency to 131. Interestingly, the 2,5-disubstituted thiophene analogue 147, with an IC50 of 2.48 μM, was considerably less potent compared to the 2,3- or 3,4-disubstituted thiophenes, 145 and 146 that showed IC50 values of 0.014 and 0.103 μM, respectively. Similar to thiophene analogues, the 2,5-disubstituted furan 148, with an IC50 of 3.57 μM, was only moderately active, while the 3,4-disubstituted analogue 149 showed comparable activity to the thiophene derivative 146. The analogue (150) bearing the aminomethyl group at the 3-position was the most potent derivative with an IC50 of 0.4 nM. Similarly, in the oxazole series, compound 153 was only moderately active (IC50 = 2.83 μM), whereas substitution at carbon adjacent to the nitrogen atom resulted in a potent compound 154 (IC50 = 7.2 nM). Nevertheless, the substitution of the aminomethyl group at the 2-position (compound 155) had a detrimental effect on the potency with an IC50 of 20 μM. Incorporation of the imidazole ring into compound 152 resulted in a weakly active compound. Furthermore, since AMD070 and related (S)-enantiomers were previously demonstrated to have a significant increase in potency, representative compounds from the pyridine 142, the thiophene 146 and the furan 149 series were synthesized as the (S)-enantiomers, resulting in compounds with an IC50 value of 4.2, 1.0, and 0.3 nM, respectively. Finally, these compounds were prepared as hydrochloride salts and were orally active in rats and dogs (Figure 16) [88].
Select examples of tetrahydroquinoline-based CXCR4 antagonists. IC50 values are the concentrations required to protect 50% MT4 cells against X4 HIV-1 NL4.3 cytopathicity [88].
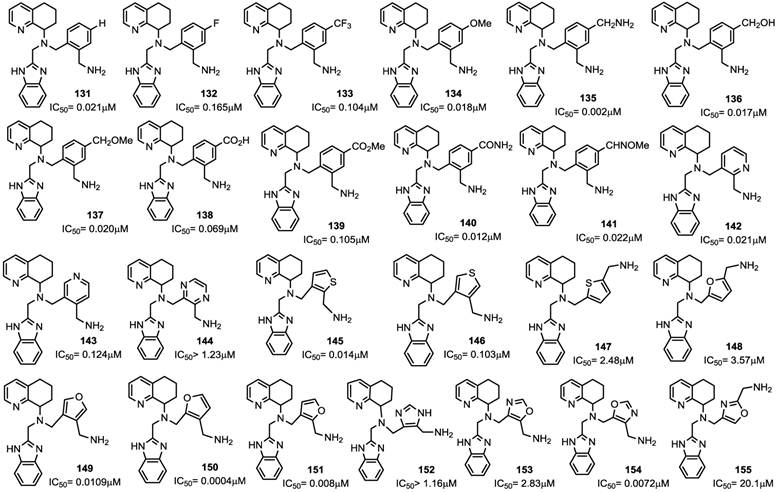
In conclusion, using AMD070 as a lead compound, several series of heterocyclic related compounds were designed and synthesized. Successful approaches in structural manipulation led to a number of compounds with antiviral activities in the low nanomolar range and acceptable pharmacokinetic properties.
Para-xylyl-enediamine-based Compounds
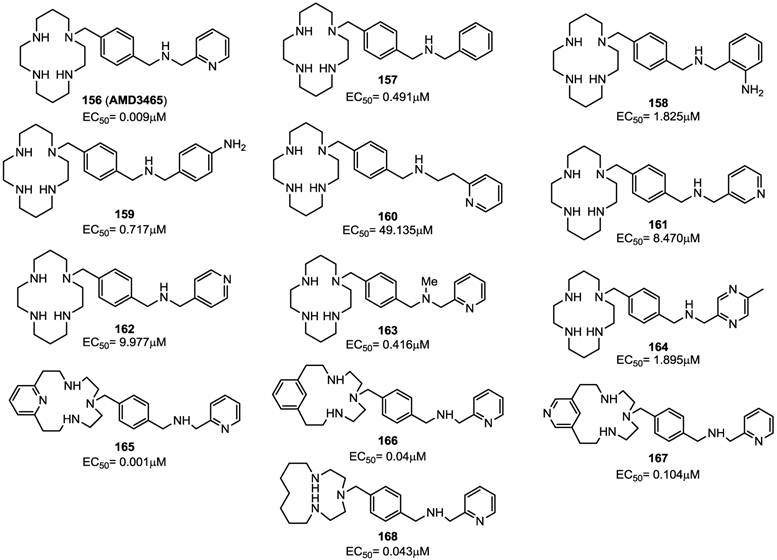
Previous efforts targeting viral entry led to the discovery of bis-tetraazamacrocycles (Bicyclams) as a class of potent and selective anti-HIV-1 agents. The prototype of such bis-macrocycles, plerixafor, was used in a recent study as a starting point for structural modification, in order to identify compounds endowed with a similar antiviral activity and improved pharmacokinetic properties [94]. In this study, the minimum structural features of plerixafor required for antiviral activity was defined by sequential replacement and/or deletion of the amino groups within the azamacrocyclic ring systems. This led to the identification of an analogue bearing a single azamacrocyclic ring AMD3465 (156) and subsequently to the design of nonmacrocyclic, orally active CXCR4 antagonists preserving a p-xylyl-enediamine linker between the two heterocyclic units of the compounds [95].
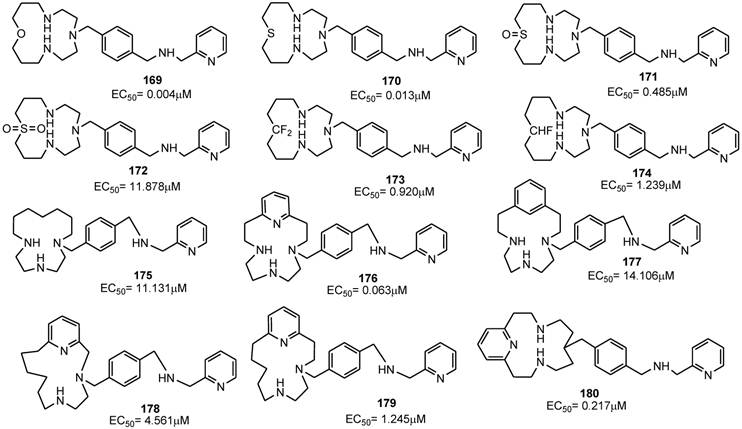
Plerixafor contains a center of symmetry and eight amino groups, four of which are positively charged at physiological pH. It was important to determine if all four positive charges were necessary for antiviral activity and to define the minimum structural requirements for activity. The replacement of one of the two tetra-aza-macrocyclic rings with a pseudo diamine-segment, mimicking the first two amino groups of the lacking macrocyclic ring from the point of attachment at the benzylic position, was attempted since it was assumed that identical features for both rings were not required. In order to avoid a second protonation at the physiological pH, a series of aminomethyl-substituted analogues, in which the second amino group was part of a heterocyclic ring or an aromatic system, were synthesized. Such compounds were tested for their inhibitory activity against a HIV-1 strain that exclusively utilizes CXCR4 for fusion and viral entry into target cells. SAR studies demonstrated that the introduction of a benzylamine group (compound 157) in place of the azamacrocyclic ring lowered the antiviral activity, although the compound showed activity with an EC50 value of 0.49 μM. Aromatic amino groups at the 2-position (compound 158) or the 4-position (compound 159) did not affect the anti-HIV activity. The replacement of the 2-pyridyl group (of compound 156) with a 3-pyridyl (compound 161) or 4-pyridyl (compound 162) led to a decreased activity of EC50(161) = 8.470 μM and EC50(162) = 9.977 μM, respectively. Also, the methylation of the amine (compound 163) reduced the anti-HIV activity. Finally, the replacement of the pyridine ring with a pyrazine moiety (compound 164) conferred an antiviral potency comparable to that of the benzyl analogue 157. As a further step, attention has been paid to the “left-hand” ring by replacing each amine groups with suitable moieties. The replacement of the cyclam ring with a py[iso-14]-ane-N4 ring led to compound 165 that showed a potent inhibitory activity with an EC50 value of 0.001 μM. Compounds 166 (containing a phenyl moiety) and 167 (containing an exocyclic-pyridine fused group), and the alkyl analog 168, retained activity (EC50 values of 0.040, 0.104 and 0.043 μM, respectively). The ether analog 169 showed to be very active too (EC50= 0.004 μM), The corresponding thioether analogue 170 exhibited an EC50 of 0.013 μM, while the sulfoxide (171) and sulfone (172) derivatives were considerably weaker antiviral agents with EC50 values of 0.485 and 11.878 μM, respectively. Furthermore, the fluoro (173) and difluoro (174) analogues showed EC50 values of 0.920 and 1.239 μM, respectively. By straightforward structural modifications, further analogues were prepared (compounds 175-180). Among them, compounds 175 and 177 showed low potency with EC50 values of 11 and 14 μM, respectively. On the other hand, compounds 176 and 180, with EC50 values of 0.063 and 0.217 μM, respectively, were active. The structures of derivatives 156-180 are shown in Figures 17 and 18 [94]. Taken together, these results suggest that there is a considerable structural nitrogen atom redundancy in plerixafor, not all amino groups are necessary for activity, and the consistence of the overall charge at physiological pH does not directly affect potency.
Representative examples of p-xylyl-enediamine based CXCR4 antagonists. EC50 values are the concentrations required to protect 50% of HIV-1 IIIB strain infected MT-4 cells against viral cytopathicity [94].
Select examples of p-xylyl-enediamine based CXCR4 antagonists. EC50 values are the concentrations required to protect 50% of HIV-1 IIIB strain infected MT-4 cells against viral cytopathicity [94].
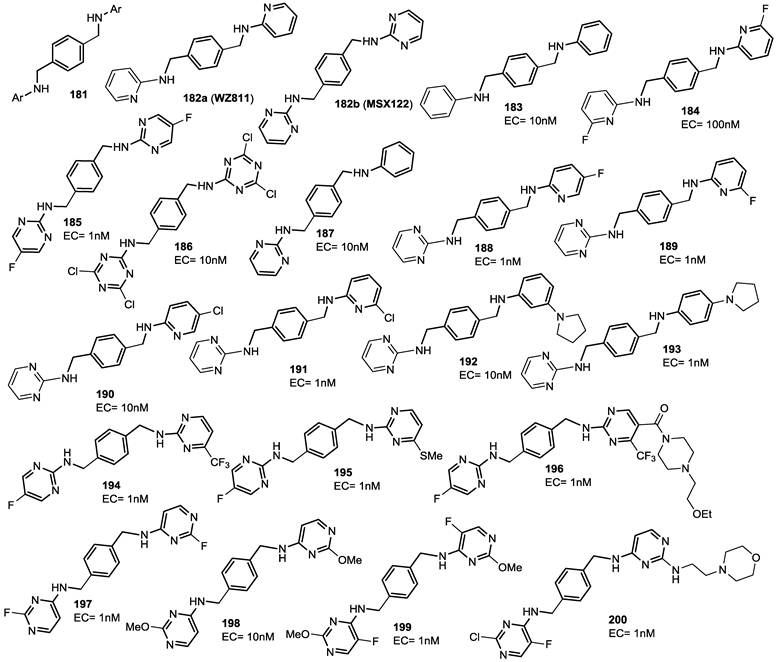
A series of dipyrimidine amines in which two heteroaromatic systems are connected by a p-xylyl-enediamine linker was already reported. These derivatives were identified through rational design and analyses of structural and pharmacological data from early reported CXCR4 inhibitors [96]. Starting from the bicyclam plerixafor and a peptidic CXCR4 antagonist (TN14003), a series of compounds, with general formula 181, were described. SAR studies indicated that the central aromatic ring is crucial for high CXCR4 affinity, and that a one-carbon separation between the central aromatic phenyl ring and the nitrogen of the acyclic linker is strictly required for potency. These results led to the identification of compounds 182a (WZ811) and 182b (MSX122) as potent CXCR4 antagonists in an affinity binding assay and in vitro functional assays. Preclinical studies suggested poor bioavailability with a consequent in vivo ineffectiveness for compound 182a [2, 97], while compound 182b entered clinical trials, unfortunately its development was soon suspended [98, 99]. Since it is plausible that the unfavorable pharmacokinetic profile of 182a is due to a rapid oxidative metabolism, a series of derivatives functionalized with electron deficient moieties on the terminal aromatic ring were synthesized. Results from binding affinity and Matrigel invasion assays (>75% at 10 nM) confirmed a CXCR4 antagonism potential for compound 185. In addition, a series of unsymmetrical compounds (187-193) with a pyrimidinyl ring at one terminus of the scaffold and a pyridinyl ring at the other side, were synthesized. Several of these derivatives showed significant activity. Furthermore, in order to optimize the pharmacokinetic profile and to improve CXCR4 binding affinity of this class of molecules, dual pyrimidinyl compounds, substituted with different functional groups such as methoxy and morpholine, were synthesized (compounds 194-200). Most of these compounds have shown potent inhibition of CXCR4 with EC values at 1 nM, with one exception, 198, EC50 value of which was 10 nM. Interestingly, all dipyrimidines displayed high potency without significant contributions from various substitutions. Compound 200 was chosen for further biological assays and showed sub-nanomolar activity in three in vitro assays including competitive binding, Matrigel invasion and GRi cyclic adenosine monophosphate (cAMP) modulation signaling. Furthermore, this compound displayed promising properties by impairing CXCR4 function in three mouse models: paw inflammation, Matrigel plug angiogenesis, and uveal melanoma micrometastasis. All these data demonstrated that dipyrimidine amines are promising CXCR4 antagonists with high potency and specificity. The structures of derivatives 181-200 are depicted in Figure 19.
Select examples of p-xylyl-enediamine based CXCR4 antagonists. EC (effective concentration) values are the concentrations at which the compound blocks > 50% of the peptide TN14003 binding to CXCR4 [96].
Guanidine-based CXCR4 Antagonists
Small molecules containing multiple guanide or biguanide groups are capable of inhibiting HIV-1 binding to CXCR4 [100]. In particular, an oligomer based on a biguanide moiety, NB325, showed wide antiviral activity in several in vitro assays. NB325 was shown to block SDF-1α signaling through CXCR4 without antagonizing the natural ligand attachment [101, 102]. However, since such preparation consists of a variable mixture of compounds containing from 4 to 10 biguanide groups, endowed with intractable activities, its development will be challenging. This study therefore focused on the design and development of a series of single small molecules containing multiple guanide or biguanide groups (Table 2, Figure 20) [100]. These compounds include some features of the polyethylene-hexamethylene biguanide NB325, as well as the peptide T140 that has five guanide groups on the side chains of arginine residues. Among the newly prepared compounds, spermidine derivatives bearing bis- and tris-phenylguanide residues, showed high affinity for CXCR4, with an IC50 of 200 nM, when compared to T140.
CXCR4-T140-fluorescein inhibitory activity of starting amines, guanide, biguanide,or phenylguanide derivatives.
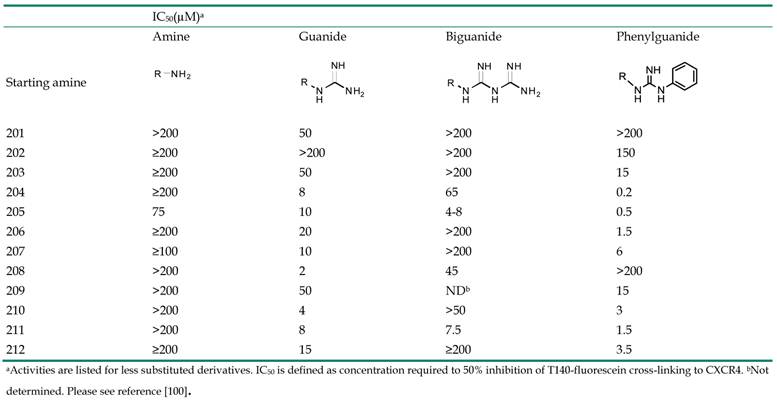
Starting amines for preparation of guanide, biguanide, or phenylguanide derivatives [100].
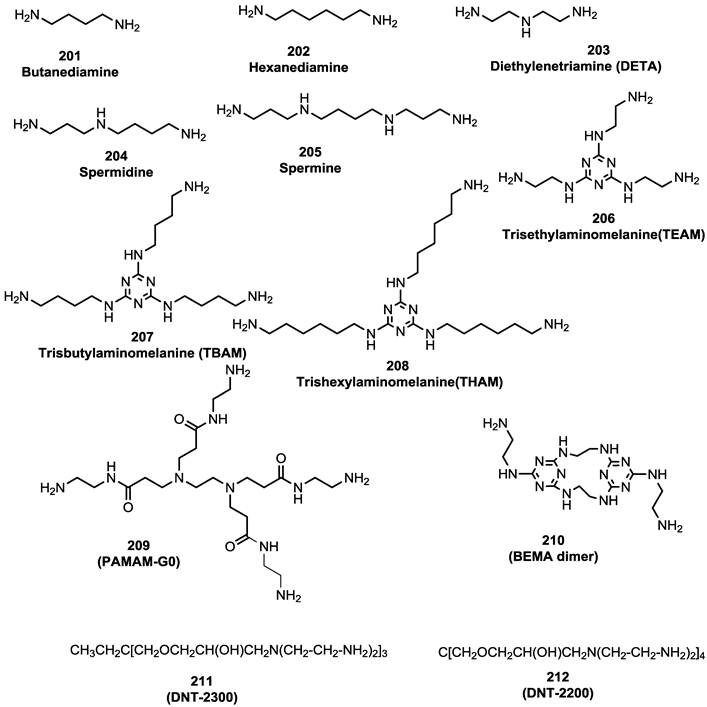
All the spermine, spermidine, and amines derivatives were tested against a panel of X4, R5, and X4/R5 isolate viral strains. Spermine phenylguanide and spermidine phenylguanide prevent the infection of X4 HIV strains but not R5 HIV strains, confirming the selectivity against CXCR4. In particular, spermidine phenylguanide showed an IC50 of 3 μM when tested against NL4-3, 92HT599, and MN X4-HIV strains. The antiviral profile was also consistent with the T140 competitive inhibition results. In these experiments, the phenylguanides showed a higher potency than the corresponding guanide or biguanide derivatives. On the other hand, the starting amines were not selective, with inhibitory activity against both X4 and R5 HIV strains. Furthermore they did not antagonize T140 binding or cross-linking to CXCR4, suggesting a different mechanism. In general, the phenylguanides were more active than either the guanides or biguanides, suggesting that an increase of the binding affinity for CXCR4 could be reached by using a larger aromatic moiety.
The positioning of the aromatic group also appeared to be important for antiviral activity. The most active derivatives of phenylguanides were characterized by the presence of aromatic groups on the main chain, have shown high binding affinity than the melamine derivative (THAM trisguanide), demonstrating the importance of the position of aromatic groups themselves. The compounds reported in this study, particularly the phenylguanides, have advantages largely because they showed higher affinities for CXCR4 and were compounds with a defined chemical composition, rather than heterogeneous mixtures such as NB325. This characteristic together with their low cytotoxicity makes them promising candidates for future in vivo evaluation to develop non-peptide CXCR4 inhibitors.
Quinoline derivatives
Recently, chloroquines and hydroxychloroquines anti-malarial drugs have been identified by computational methods as promising inhibitors of the binding of SDF1-α to CXCR4 and thus, potential agents for certain forms of cancer [103-106]. Further studies have focused on the anti-neoplastic effects of chloroquine and hydroxychloroquine in the treatment of colon and pancreatic cancers although their mechanisms of action maybe due to their effects on multiple pathways [107-109].
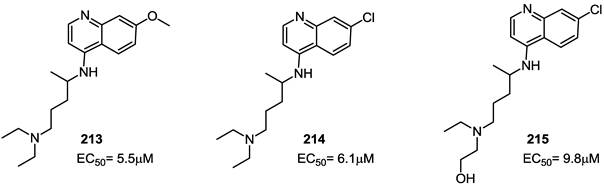
Using the predicted conformations of CXCR4, libraries of compounds have been screened to identify new antagonists [103]. Among the top candidates, compound 213 (NSC56612) was selected as the lead compound by high-throughput screening. The inspection of several databases led to the identification of structural similarity between such lead and numerous quinoline or acridine derivatives, such as chloroquine, hydroxychloroquine and quinacrine. In order to identify the binding sites of these derivatives, chloroquine 214 and hydroxychloroquine 215 were docked onto to the crystal structure of CXCR4. The results suggested that the tertiary amine groups bind to D97 on TM2 and E32 in the amino terminus of CXCR4 through hydrogen bonds while aromatic residues Y45, W94, H113 and Y255 establish hydrophobic interactions with the aromatic counterpart of identified compounds.
When tested in in vitro assays, compounds 213, 214 and 215 showed EC50 values of 5.5, 6.1 and 9.8 μM, respectively (Figure 21). Furthermore, since earlier studies have established the crucial role of MAPK in mediating growth and proliferation in pancreatic intraepithelial neoplasia and pancreatic cancer cells, these derivatives were used to evaluate the MAPK signaling pathway. Effective inhibition of SDF-1-driven ERK phosphorylation and inhibition of SDF-1-mediated in vitro proliferation were observed.
Structure of quinoline based CXCR4 antagonists. EC50 values are the concentrations for 50% inhibition of the SDF-1α binding to CXCR4 receptor in HEK293 cells [103].
Miscellaneous Compounds
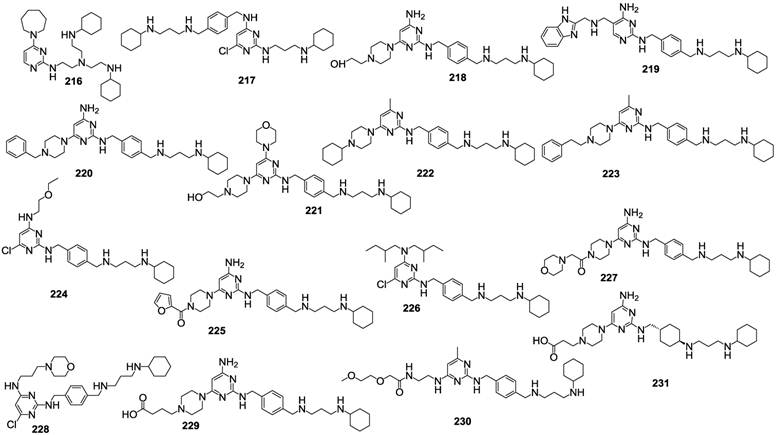
Numerous recent articles, including some patent applications, deal with the identification of compounds belonging to diverse chemical classes, claiming to inhibit CXCR4, thus, potentially useful for a broad clinical application. For example, a recent patent application provided a series of variously substituted pyrimidines claimed as CXCR4 antagonists. A few representative examples out of the 268 described compounds are shown in Figure 22 [110]. 196 of the tested compounds showed IC50 values in the range of 0.003-0.1 μM, 56 in the range of 0.1-1 μM and 16 in the range of 1-5 μM, suggesting that such class of derivatives is worthy of further development.
Representative examples of pyrimidine-based CXCR4 antagonists [110].
A separate patent application deals with compounds whose general formula is reported in Figure 23 [111]. Such compounds are claimed useful for the treatment of several diseases involving a CXCR4 biological role. A recent work reported additional N-containing scaffolds as novel CXCR4 antagonists [112].
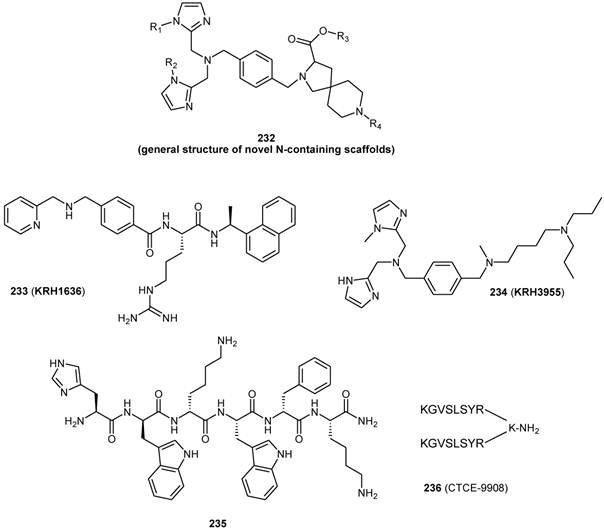
Previously, a low molecular weight non-peptide compound 233, KRH-1636, was found to be a potent and selective inhibitor of CXCR4 using X4 HIV-1 strains, but, its poor oral bioavailability limited further clinical trials [113, 114]. A newly developed KRH-1636 derivative, compound 234 (KRH-3955), was instead reported to be orally active. This compound potently inhibit the X4 HIV-1 replication within different clinical studies. KRH-3955 inhibits both SDF-1α and anti-CXCR4 mAB binding to CXCR4, as well as Ca2+ signaling through the receptor [113].
A recent paper described the identification of a ghrelin receptor blocker, compound 235 (D-[Lys3] GHRP-6) as a CXCR4 antagonist [115]. The functions of Growth Hormone Secretagogue Receptor (GHS-R) were formerly assessed by studies carried out with synthetic peptidyl agonists. Among them, the most useful was demonstrated to be the Growth Hormone Releasing Peptide-6 (GHRP-6), before the discovery of the endogenous ligand ghrelin. The modification of GHRP-6 (H-His-D-Trp-Ala-Trp-D-Phe-Lys-NH2) from alanine to D-lysine led to a GHS-R antagonist, D-[Lys3]GHRP-6 (235), widely used today as a selective GHS-R antagonist in recent studies [116, 117]. Compound 235 partially inhibits CXCR4 functions and signaling. Structural analogues with similar binding properties may also be active against HIV infection, cancer cell migration, and inflammatory disorders mediated by CXCR4.
Several modified peptides were discovered as CXCR4 antagonists. Chemokine Therapeutics Corporation's CTCE-9908 (236) and Polyphor Ltd's POL6326 (structure not disclosed publicly) are few examples of these modified peptides [118-120]. CTCE-9908 is a peptide with 17 amino acid sequence, which is a dimer of the 8 amino acid N-terminal sequence with modified P to G, bridges by lysine [118, 119]. POL6326 is designed through protein epitope mimetic (PEM) approach, where epitope from a recombinant protein is transferred to a synthetic scaffold [120].
CXCR4 Antagonists in Clinical Trials
Currently, only one CXCR4 antagonist, plerixafor is approved by the FDA and several others are in clinical trials (Table 3). Plerixafor in combination with granulocyte-colony stimulating factor (G-CSF) has been approved for hematopoietic stem cell mobilization to the peripheral blood in patients with non-Hodgkin's lymphoma and multiple myeloma [54]. Also, it is currently in clinical trials in combination with other chemotherapeutic agents for the prevention of growth and metastasis of a number of cancers including glioma [121], acute myeloid leukemia (AML) [122], chronic lymphocytic leukemia (CLL) [123]. Plerixafor is in a phase I clinical trial against myelokathexis (WHIM syndrome) [124]. Another CXCR4 antagonist, TG-0054 is in phase II clinical study for stem cell mobilization in patients with multiple myeloma, non-Hodgkin lymphoma or Hodgkin disease [125]. The orally available CXCR4 antagonist, AMD070 has been studied in a phase I/II clinical trial in HIV infected adults with X4-tropic virus [126]. Although the phase I/II study of AMD070 has been completed in January 2009, there is no further news about its development. Previously, the orally active CXCR4 antagonist, MSX-122, was tested in a phase I clinical trial against solid tumors, but in 2008 the study was suspended due to undisclosed reasons [127]. CTCE-9908 is in phase I/II clinical trial against patients with advanced solid tumors [98]. There is no further news on advancement of clinical trial although phase I/II trial has been completed in 2008. Another modified peptide POL6326 is in phase II clinical trial in hematopoietic stem cell mobilization in multiple myeloma [128].
CXCR4 antagonists under clinical investigation.
| Agents | Phase | Indication | Sponsor |
|---|---|---|---|
| Plerixafor | FDA approved | Hematopoetic stem cell mobilization in patients with non-Hodgkin's lymphoma and multiple myeloma | Genozyme |
| Plerixafor | Phase I | Glioma, Acute Myeloid Leukemia, Chronic Lymphocytic Leukemia | Genozyme |
| Plerixafor | Phase I | Myelokathexis (WHIM syndrome) | Genozyme |
| TG-0054 | Phase II | Hematopoietic stem cell mobilization in patients with multiple myeloma, non-Hodgkin lymphoma or Hodgkin disease | TaiGen Biotechnology Co., Ltd. |
| AMD070 | Phase I/II | HIV Infections | National Institute of Allergy and Infectious Diseases (NIAID) |
| MSX-122 | Phase I | Refractory Metastatic or Locally Advanced Solid Tumors | Metastatix, Inc. |
| CTCE-9908 | Phase I/II | Advanced Solid Tumors | Chemokine Therapeutics Corp. |
| POL6326 | Phase II | Hematopoetic stem cell mobilization in patients with Leukemia, Lymphoma and Multiple Myeloma | Polyphor Ltd. |
Conclusions
The diverse role of the SDF-1/CXCR4 axis in the pathogenesis of HIV, cancer, and rheumatoid arthritis, has established CXCR4 as a promising therapeutic target. Considering the vast population of patients with these diseases, the development of CXCR4 antagonists is of therapeutic as well as economic importance. Currently, only one CXCR4 antagonist, plerixafor, has been approved by the FDA for hematopoietic stem cell mobilization as an injectable agent for short-term treatments. Novel CXCR4 antagonists, including the orally active AMD70, are currently in clinical trials for various indications. Since AIDS is a chronic disease, CXCR4 antagonists that are orally available and safe for long-term use, are in medical need for the treatment of HIV infection. While the long-term toxicity and off-target effects of early stage candidate drugs need to be fully characterized, the development of safe and orally active CXCR4 antagonists is a major goal for medicinal chemists. The diverse set of antagonists described in this article will be useful in guiding medicinal chemists to rationally design effective CXCR4 antagonists for the treatment of human diseases where SDF-1/CXCR4 axis plays a critical role.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lefrancois M, Lefebvre MR, Saint-Onge G, Boulais PE, Lamothe S, Leduc R. et al. Agonists for the Chemokine Receptor CXCR4. ACS Med Chem Lett. 2011;2:597-602
2. Zhan W, Liang Z, Zhu A, Kurtkaya S, Shim H, Snyder JP. et al. Discovery of small molecule CXCR4 antagonists. J Med Chem. 2007;50:5655-64
3. Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci U S A. 1997;94:1925-30
4. Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F. et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833-5
5. Bleul CC, Farzan M, Choe H, Parolin C, Clark-Lewis I, Sodroski J. et al. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature. 1996;382:829-33
6. Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787-820
7. Raman D, Sobolik-Delmaire T, Richmond A. Chemokines in health and disease. Exp Cell Res. 2011;317:575-89
8. Balkwill FR. The chemokine system and cancer. J Pathol. 2012;226:148-57
9. Fagete S, Ravn U, Gueneau F, Magistrelli G, Kosco-Vilbois MH, Fischer N. Specificity tuning of antibody fragments to neutralize two human chemokines with a single agent. MAbs. 2009;1:288-96
10. Woodard LE, Nimmagadda S. CXCR4-based imaging agents. J Nucl Med. 2011;52:1665-9
11. Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V. et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066-71
12. Crump MP, Gong JH, Loetscher P, Rajarathnam K, Amara A, Arenzana-Seisdedos F. et al. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. Embo J. 1997;16:6996-7007
13. Didigu CA, Doms RW. Novel Approaches to Inhibit HIV Entry. Viruses. 2012;4:309-24
14. Azevedo-Pereira JM, Santos-Costa Q. [Chemokine receptors and its importance in the replication cycle of human immunodeficiency virus: clinical and therapeutic implications]. Acta Med Port. 2008;21:497-504
15. Seibert C, Sakmar TP. Small-molecule antagonists of CCR5 and CXCR4: a promising new class of anti-HIV-1 drugs. Curr Pharm Des. 2004;10:2041-62
16. Cervia JS, Smith MA. Enfuvirtide (T-20): a novel human immunodeficiency virus type 1 fusion inhibitor. Clin Infect Dis. 2003;37:1102-6
17. Grande F, Garofalo A, Neamati N. Small molecules anti-HIV therapeutics targeting CXCR4. Curr Pharm Des. 2008;14:385-404
18. Wilen CB, Tilton JC, Doms RW. Molecular mechanisms of HIV entry. Adv Exp Med Biol. 2012;726:223-42
19. Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med. 1997;185:621-8
20. Sucupira MC, Sanabani S, Cortes RM, Giret MT, Tomiyama H, Sauer MM. et al. Faster HIV-1 disease progression among Brazilian individuals recently infected with CXCR4-utilizing strains. PLoS One. 2012;7:e30292
21. Raymond S, Delobel P, Mavigner M, Cazabat M, Encinas S, Souyris C. et al. CXCR4-using viruses in plasma and peripheral blood mononuclear cells during primary HIV-1 infection and impact on disease progression. Aids. 2010;24:2305-12
22. Furusato B, Mohamed A, Uhlen M, Rhim JS. CXCR4 and cancer. Pathol Int. 2010;60:497-505
23. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. 1999;94:3658-67
24. Mohle R, Failenschmid C, Bautz F, Kanz L. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1). Leukemia. 1999;13:1954-9
25. Burger M, Hartmann T, Krome M, Rawluk J, Tamamura H, Fujii N. et al. Small peptide inhibitors of the CXCR4 chemokine receptor (CD184) antagonize the activation, migration, and antiapoptotic responses of CXCL12 in chronic lymphocytic leukemia B cells. Blood. 2005;106:1824-30
26. Bradstock KF, Makrynikola V, Bianchi A, Shen W, Hewson J, Gottlieb DJ. Effects of the chemokine stromal cell-derived factor-1 on the migration and localization of precursor-B acute lymphoblastic leukemia cells within bone marrow stromal layers. Leukemia. 2000;14:882-8
27. Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523-30
28. Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
29. Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M. et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell. 2004;6:459-69
30. Burger M, Glodek A, Hartmann T, Schmitt-Graff A, Silberstein LE, Fujii N. et al. Functional expression of CXCR4 (CD184) on small-cell lung cancer cells mediates migration, integrin activation, and adhesion to stromal cells. Oncogene. 2003;22:8093-101
31. Kijima T, Maulik G, Ma PC, Tibaldi EV, Turner RE, Rollins B. et al. Regulation of cellular proliferation, cytoskeletal function, and signal transduction through CXCR4 and c-Kit in small cell lung cancer cells. Cancer Res. 2002;62:6304-11
32. Wang J, Sun Y, Song W, Nor JE, Wang CY, Taichman RS. Diverse signaling pathways through the SDF-1/CXCR4 chemokine axis in prostate cancer cell lines leads to altered patterns of cytokine secretion and angiogenesis. Cell Signal. 2005;17:1578-92
33. Barbolina MV, Kim M, Liu Y, Shepard J, Belmadani A, Miller RJ. et al. Microenvironmental regulation of chemokine (C-X-C-motif) receptor 4 in ovarian carcinoma. Mol Cancer Res. 2010;8:653-64
34. Zeelenberg IS, Ruuls-Van Stalle L, Roos E. The chemokine receptor CXCR4 is required for outgrowth of colon carcinoma micrometastases. Cancer Res. 2003;63:3833-9
35. Burger JA. Chemokines and chemokine receptors in chronic lymphocytic leukemia (CLL): from understanding the basics towards therapeutic targeting. Semin Cancer Biol. 2010;20:424-30
36. Liang Z, Yoon Y, Votaw J, Goodman MM, Williams L, Shim H. Silencing of CXCR4 blocks breast cancer metastasis. Cancer Res. 2005;65:967-71
37. Li RJ, Zhao LJ, Zhan ZL, Lu X, Gong LL, Wang P. [Significance of expression of chemokine receptor and matrix metalloproteinase in small cell lung cancer]. Zhonghua Yi Xue Za Zhi. 2012;92:532-5
38. Gladson CL, Welch DR. New insights into the role of CXCR4 in prostate cancer metastasis. Cancer Biol Ther. 2008;7:1849-51
39. Akashi T, Koizumi K, Tsuneyama K, Saiki I, Takano Y, Fuse H. Chemokine receptor CXCR4 expression and prognosis in patients with metastatic prostate cancer. Cancer Sci. 2008;99:539-42
40. Kim M, Koh YJ, Kim KE, Koh BI, Nam DH, Alitalo K. et al. CXCR4 signaling regulates metastasis of chemoresistant melanoma cells by a lymphatic metastatic niche. Cancer Res. 2010;70:10411-21
41. Franco R, Botti G, Mascolo M, Loquercio G, Liguori G, Ilardi G. et al. "CXCR4-CXCL12 and VEGF correlate to uveal melanoma progression". Front Biosci (Elite Ed). 2010;2:13-21
42. Chung SH, Seki K, Choi BI, Kimura KB, Ito A, Fujikado N. et al. CXC chemokine receptor 4 expressed in T cells plays an important role in the development of collagen-induced arthritis. Arthritis Res Ther. 2010;12:R188
43. De Klerck B, Geboes L, Hatse S, Kelchtermans H, Meyvis Y, Vermeire K. et al. Pro-inflammatory properties of stromal cell-derived factor-1 (CXCL12) in collagen-induced arthritis. Arthritis Res Ther. 2005;7:R1208-20
44. Buckley CD, Amft N, Bradfield PF, Pilling D, Ross E, Arenzana-Seisdedos F. et al. Persistent induction of the chemokine receptor CXCR4 by TGF-beta 1 on synovial T cells contributes to their accumulation within the rheumatoid synovium. J Immunol. 2000;165:3423-9
45. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F. et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003;34:70-4
46. Bachelerie F. CXCL12/CXCR4-axis dysfunctions: Markers of the rare immunodeficiency disorder WHIM syndrome. Dis Markers. 2010;29:189-98
47. Wen J, Zhang JQ, Huang W, Wang Y. SDF-1alpha and CXCR4 as therapeutic targets in cardiovascular disease. Am J Cardiovasc Dis. 2012;2:20-8
48. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858-64
49. Misao Y, Takemura G, Arai M, Ohno T, Onogi H, Takahashi T. et al. Importance of recruitment of bone marrow-derived CXCR4+ cells in post-infarct cardiac repair mediated by G-CSF. Cardiovasc Res. 2006;71:455-65
50. Song JS, Kang CM, Kang HH, Yoon HK, Kim YK, Kim KH. et al. Inhibitory effect of CXC chemokine receptor 4 antagonist AMD3100 on bleomycin induced murine pulmonary fibrosis. Exp Mol Med. 2010;42:465-72
51. Wang A, Guilpain P, Chong BF, Chouzenoux S, Guillevin L, Du Y. et al. Dysregulated expression of CXCR4/CXCL12 in subsets of patients with systemic lupus erythematosus. Arthritis Rheum. 2010;62:3436-46
52. Chong BF, Mohan C. Targeting the CXCR4/CXCL12 axis in systemic lupus erythematosus. Expert Opin Ther Targets. 2009;13:1147-53
53. Wang A, Fairhurst AM, Tus K, Subramanian S, Liu Y, Lin F. et al. CXCR4/CXCL12 hyperexpression plays a pivotal role in the pathogenesis of lupus. J Immunol. 2009;182:4448-58
54. Burger JA, Stewart DJ. CXCR4 chemokine receptor antagonists: perspectives in SCLC. Expert Opin Investig Drugs. 2009;18:481-90
55. Harvey JR, Mellor P, Eldaly H, Lennard TW, Kirby JA, Ali S. Inhibition of CXCR4-mediated breast cancer metastasis: a potential role for heparinoids? Clin Cancer Res. 2007;13:1562-70
56. Wilson LJ, Liotta DC. Emergence of small-molecule CXCR4 antagonists as novel immune and hematopoietic system regulatory agents. Drug Development Research. 2011;72:598-602
57. Tsibris AM, Kuritzkes DR. Chemokine antagonists as therapeutics: focus on HIV-1. Annu Rev Med. 2007;58:445-59
58. Tamamura H, Fujii N. The therapeutic potential of CXCR4 antagonists in the treatment of HIV infection, cancer metastasis and rheumatoid arthritis. Expert Opin Ther Targets. 2005;9:1267-82
59. Fujii N, Nakashima H, Tamamura H. The therapeutic potential of CXCR4 antagonists in the treatment of HIV. Expert Opin Investig Drugs. 2003;12:185-95
60. Zhou N, Luo Z, Luo J, Fan X, Cayabyab M, Hiraoka M. et al. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with D-peptides derived from chemokines. J Biol Chem. 2002;277:17476-85
61. Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA. et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667-73
62. Tanaka T, Nomura W, Narumi T, Esaka A, Oishi S, Ohashi N. et al. Structure-activity relationship study on artificial CXCR4 ligands possessing the cyclic pentapeptide scaffold: the exploration of amino acid residues of pentapeptides by substitutions of several aromatic amino acids. Org Biomol Chem. 2009;7:3805-9
63. Tanaka T, Tsutsumi H, Nomura W, Tanabe Y, Ohashi N, Esaka A. et al. Structure-activity relationship study of CXCR4 antagonists bearing the cyclic pentapeptide scaffold: identification of the new pharmacophore. Org Biomol Chem. 2008;6:4374-7
64. Tamamura H, Esaka A, Ogawa T, Araki T, Ueda S, Wang Z. et al. Structure-activity relationship studies on CXCR4 antagonists having cyclic pentapeptide scaffolds. Org Biomol Chem. 2005;3:4392-4
65. Fujii N, Oishi S, Hiramatsu K, Araki T, Ueda S, Tamamura H. et al. Molecular-size reduction of a potent CXCR4-chemokine antagonist using orthogonal combination of conformation- and sequence-based libraries. Angew Chem Int Ed Engl. 2003;42:3251-3
66. Ueda S, Oishi S, Wang ZX, Araki T, Tamamura H, Cluzeau J. et al. Structure-activity relationships of cyclic peptide-based chemokine receptor CXCR4 antagonists: disclosing the importance of side-chain and backbone functionalities. J Med Chem. 2007;50:192-8
67. Tamamura H, Araki T, Ueda S, Wang Z, Oishi S, Esaka A. et al. Identification of novel low molecular weight CXCR4 antagonists by structural tuning of cyclic tetrapeptide scaffolds. J Med Chem. 2005;48:3280-9
68. Tamamura H, Hiramatsu K, Ueda S, Wang Z, Kusano S, Terakubo S. et al. Stereoselective synthesis of [L-Arg-L/D-3-(2-naphthyl)alanine]-type (E)-alkene dipeptide isosteres and its application to the synthesis and biological evaluation of pseudopeptide analogues of the CXCR4 antagonist FC131. J Med Chem. 2005;48:380-91
69. Narumi T, Hayashi R, Tomita K, Kobayashi K, Tanahara N, Ohno H. et al. Synthesis and biological evaluation of selective CXCR4 antagonists containing alkene dipeptide isosteres. Org Biomol Chem. 2010;8:616-21
70. Inokuchi E, Oishi S, Kubo T, Ohno H, Shimura K, Matsuoka M. et al. Potent CXCR4 antagonists containing amidine type peptide bond isosteres. ACS Med Chem Lett. 2011;2:477-80
71. Oishi S, Kamitani H, Kodera Y, Watanabe K, Kobayashi K, Narumi T. et al. Peptide bond mimicry by (E)-alkene and (Z)-fluoroalkene peptide isosteres: synthesis and bioevaluation of alpha-helical anti-HIV peptide analogues. Org Biomol Chem. 2009;7:2872-7
72. Kobayashi K, Narumi T, Oishi S, Ohno H, Fujii N. Amino acid-based synthesis of trifluoromethylalkene dipeptide isosteres by alcohol-assisted nucleophilic trifluoromethylation and organozinc-copper-mediated S(N)2' alkylation. J Org Chem. 2009;74:4626-9
73. Yamaki Y, Shigenaga A, Li J, Shimohigashi Y, Otaka A. Synthesis of amide-type fluoroalkene dipeptide isosteres by an intramolecular redox reaction. J Org Chem. 2009;74:3278-85
74. Dai N, Wang XJ, Etzkorn FA. The effect of a trans-locked Gly-Pro alkene isostere on collagen triple helix stability. J Am Chem Soc. 2008;130:5396-7
75. Wiktelius D, Luthman K. Taking control of P1, P1' and double bond stereochemistry in the synthesis of Phe-Phe (E)-alkene amide isostere dipeptidomimetics. Org Biomol Chem. 2007;5:603-5
76. Bandur NG, Harms K, Koert U. First stereoselective snthesis of a Tyr-Tyr E-alkene isostere. Synlett. 2007:99-102
77. Jones RCF, Ward GJ. Amide bond isosteres: imidazolines in pseudopeptide chemistry. Tetrahedron Lett. 1988;29:3853-6
78. Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed Engl. 2006;45:2348-68
79. Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Curr Opin Chem Biol. 2000;4:696-703
80. Li ZB, Chen K, Chen X. (68)Ga-labeled multimeric RGD peptides for microPET imaging of integrin alpha(v)beta (3) expression. Eur J Nucl Med Mol Imaging. 2008;35:1100-8
81. Demmer O, Dijkgraaf I, Schumacher U, Marinelli L, Cosconati S, Gourni E. et al. Design, synthesis, and functionalization of dimeric peptides targeting chemokine receptor CXCR4. J Med Chem. 2011;54:7648-62
82. Tanaka T, Nomura W, Narumi T, Masuda A, Tamamura H. Bivalent ligands of CXCR4 with rigid linkers for elucidation of the dimerization state in cells. J Am Chem Soc. 2010;132:15899-901
83. Demmer O, Gourni E, Schumacher U, Kessler H, Wester HJ. PET imaging of CXCR4 receptors in cancer by a new optimized ligand. ChemMedChem. 2011;6:1789-91
84. Ueda S, Kato M, Inuki S, Ohno H, Evans B, Wang ZX. et al. Identification of novel non-peptide CXCR4 antagonists by ligand-based design approach. Bioorg Med Chem Lett. 2008;18:4124-9
85. Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E. et al. Pharmacokinetics and safety of AMD-3100, a novel antagonist of the CXCR-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44:1667-73
86. Moyle G, DeJesus E, Boffito M, Wong RS, Gibney C, Badel K. et al. Proof of activity with AMD11070, an orally bioavailable inhibitor of CXCR4-tropic HIV type 1. Clin Infect Dis. 2009;48:798-805
87. Skerlj R, Bridger G, McEachern E, Harwig C, Smith C, Kaller A. et al. Design of novel CXCR4 antagonists that are potent inhibitors of T-tropic (X4) HIV-1 replication. Bioorg Med Chem Lett. 2011;21:1414-8
88. Skerlj R, Bridger G, McEachern E, Harwig C, Smith C, Wilson T. et al. Synthesis and SAR of novel CXCR4 antagonists that are potent inhibitors of T tropic (X4) HIV-1 replication. Bioorg Med Chem Lett. 2011;21:262-6
89. Miller JF, Gudmundsson KS, D'Aurora Richardson L, Jenkinson S, Spaltenstein A, Thomson M. et al. Synthesis and SAR of novel isoquinoline CXCR4 antagonists with potent anti-HIV activity. Bioorg Med Chem Lett. 2010;20:3026-30
90. Miller JF, Turner EM, Gudmundsson KS, Jenkinson S, Spaltenstein A, Thomson M. et al. Novel N-substituted benzimidazole CXCR4 antagonists as potential anti-HIV agents. Bioorg Med Chem Lett. 2010;20:2125-8
91. Catalano JG, Gudmundsson KS, Svolto A, Boggs SD, Miller JF, Spaltenstein A. et al. Synthesis of a novel tricyclic 1,2,3,4,4a,5,6,10b-octahydro-1,10-phenanthroline ring system and CXCR4 antagonists with potent activity against HIV-1. Bioorg Med Chem Lett. 2010;20:2186-90
92. Gudmundsson KS, Boggs SD, Catalano JG, Svolto A, Spaltenstein A, Thomson M. et al. Imidazopyridine-5,6,7,8-tetrahydro-8-quinolinamine derivatives with potent activity against HIV-1. Bioorg Med Chem Lett. 2009;19:6399-403
93. Gudmundsson KS, Sebahar PR, Richardson LD, Miller JF, Turner EM, Catalano JG. et al. Amine substituted N-(1H-benzimidazol-2ylmethyl)-5,6,7,8-tetrahydro-8-quinolinamines as CXCR4 antagonists with potent activity against HIV-1. Bioorg Med Chem Lett. 2009;19:5048-52
94. Bridger GJ, Skerlj RT, Hernandez-Abad PE, Bogucki DE, Wang Z, Zhou Y. et al. Synthesis and structure-activity relationships of azamacrocyclic C-X-C chemokine receptor 4 antagonists: analogues containing a single azamacrocyclic ring are potent inhibitors of T-cell tropic (X4) HIV-1 replication. J Med Chem. 2010;53:1250-60
95. Bodart V, Anastassov V, Darkes MC, Idzan SR, Labrecque J, Lau G. et al. Pharmacology of AMD3465: a small molecule antagonist of the chemokine receptor CXCR4. Biochem Pharmacol. 2009;78:993-1000
96. Zhu A, Zhan W, Liang Z, Yoon Y, Yang H, Grossniklaus HE. et al. Dipyrimidine amines: a novel class of chemokine receptor type 4 antagonists with high specificity. J Med Chem. 2010;53:8556-68
97. Fujii N, Tamamura H. Peptide-lead CXCR4 antagonists with high anti-HIV activity. Curr Opin Investig Drugs. 2001;2:1198-202
98. Wong D, Korz W. Translating an Antagonist of Chemokine Receptor CXCR4: from bench to bedside. Clin Cancer Res. 2008;14:7975-80
99. Yu L, Cecil J, Peng SB, Schrementi J, Kovacevic S, Paul D. et al. Identification and expression of novel isoforms of human stromal cell-derived factor 1. Gene. 2006;374:174-9
100. Wilkinson RA, Pincus SH, Shepard JB, Walton SK, Bergin EP, Labib M. et al. Novel compounds containing multiple guanide groups that bind the HIV coreceptor CXCR4. Antimicrob Agents Chemother. 2011;55:255-63
101. Thakkar N, Pirrone V, Passic S, Zhu W, Kholodovych V, Welsh W. et al. Specific interactions between the viral coreceptor CXCR4 and the biguanide-based compound NB325 mediate inhibition of human immunodeficiency virus type 1 infection. Antimicrob Agents Chemother. 2009;53:631-8
102. Krebs FC, Miller SR, Ferguson ML, Labib M, Rando RF, Wigdahl B. Polybiguanides, particularly polyethylene hexamethylene biguanide, have activity against human immunodeficiency virus type 1. Biomed Pharmacother. 2005;59:438-45
103. Kim J, Yip ML, Shen X, Li H, Hsin LY, Labarge S. et al. Identification of anti-malarial compounds as novel antagonists to chemokine receptor CXCR4 in pancreatic cancer cells. PLoS One. 2012;7:e31004
104. Ben-Zvi I, Kivity S, Langevitz P, Shoenfeld Y. Hydroxychloroquine: from malaria to autoimmunity. Clin Rev Allergy Immunol. 2012;42:145-53
105. Hempelmann E. Hemozoin biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors. Parasitol Res. 2007;100:671-6
106. Warhurst DC, Steele JC, Adagu IS, Craig JC, Cullander C. Hydroxychloroquine is much less active than chloroquine against chloroquine-resistant Plasmodium falciparum, in agreement with its physicochemical properties. J Antimicrob Chemother. 2003;52:188-93
107. Sasaki K, Tsuno NH, Sunami E, Tsurita G, Kawai K, Okaji Y. et al. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer. 2010;10:370
108. Di Marco M, Di Cicilia R, Macchini M, Nobili E, Vecchiarelli S, Brandi G. et al. Metastatic pancreatic cancer: is gemcitabine still the best standard treatment? (Review). Oncol Rep. 2010;23:1183-92
109. Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S. et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960-6
110. Yen CF, Hu CK, Chou MC, Tseng CT, Wu CH, Huang YH, Chen SJ, King CHR. Pyrimidine compounds. US 2009/0143302 (A1). 2009
111. Yoshida K, Ochiai H, Tani K, Shibayama S, Kasano M. Compound having spiro-bonded cyclic group and use thereof. WO 2010/147094 (A1). 2010
112. De Clercq E. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther. 2010;128:509-18
113. Murakami T, Kumakura S, Yamazaki T, Tanaka R, Hamatake M, Okuma K. et al. The novel CXCR4 antagonist KRH-3955 is an orally bioavailable and extremely potent inhibitor of human immunodeficiency virus type 1 infection: comparative studies with AMD3100. Antimicrob Agents Chemother. 2009;53:2940-8
114. Ichiyama K, Yokoyama-Kumakura S, Tanaka Y, Tanaka R, Hirose K, Bannai K. et al. A duodenally absorbable CXC chemokine receptor 4 antagonist, KRH-1636, exhibits a potent and selective anti-HIV-1 activity. Proc Natl Acad Sci U S A. 2003;100:4185-90
115. Patel K, Dixit VD, Lee JH, Kim JW, Schaffer EM, Nguyen D. et al. Identification of ghrelin receptor blocker, D-[Lys3] GHRP-6 as a CXCR4 receptor antagonist. Int J Biol Sci. 2012;8:108-17
116. Smith RG. Development of growth hormone secretagogues. Endocr Rev. 2005;26:346-60
117. Kojima M, Kangawa K. Ghrelin: structure and function. Physiol Rev. 2005;85:495-522
118. Hassan S, Buchanan M, Jahan K, Aguilar-Mahecha A, Gaboury L, Muller WJ. et al. CXCR4 peptide antagonist inhibits primary breast tumor growth, metastasis and enhances the efficacy of anti-VEGF treatment or docetaxel in a transgenic mouse model. Int J Cancer. 2011;129:225-32
119. Huang EH, Singh B, Cristofanilli M, Gelovani J, Wei C, Vincent L. et al. A CXCR4 antagonist CTCE-9908 inhibits primary tumor growth and metastasis of breast cancer. J Surg Res. 2009;155:231-6
120. Robinson JA, Demarco S, Gombert F, Moehle K, Obrecht D. The design, structures and therapeutic potential of protein epitope mimetics. Drug Discov Today. 2008;13:944-51
121. Plerixafor (AMD3100) and Bevacizumab for Recurrent High-Grade Glioma. NCT01339039. http://clinicaltrialsgov
122. Feasibility and Efficiency Study of Leukemic Cell Mobilization With Plerixafor Injection. NCT01141543. http://clinicaltrialsgov
123. Lenalidomide + Plerixafor in Previously Treated Chronic Lymphocytic Leukemia (CLL). NCT01373229. http://clinicaltrialsgov
124. AMD 3100 for Treatment of Myelokathexis. NCT01058993. http://clinicaltrialsgov
125. Safety, Pharmacokinetics and Stem Cell Mobilization of TG-0054 in Multiple Myeloma, Non-Hodgkin Lymphoma and Hodgkin Disease Patients. NCT01018979. http://clinicaltrialsgov
126. Safety and Activity of the Oral HIV Entry Inhibitor AMD11070 in HIV Infected Patients. NCT00089466. http://clinicaltrialsgov
127. MSX-122 Administered Orally in Patients With Refractory Metastatic or Locally Advanced Solid Tumors. NCT00591682. http://clinicaltrialsgov
128. Safety and Efficacy of POL6326 for Mobilization/Transplant of Sibling Donor in Patients With Hematologic Malignancies. NCT01413568. http://clinicaltrialsgov
129. Tanguy Le Carrour SA, Sylvie Tondeur, Ludovic Lhermitte, Ned Lamb, Thierry Reme, Veronique Pantesco, Samir Hamamah, Bernard Klein, John De Vos. Amazonia!: an online resource to google and visualize public human whole genome expression data. The open bioinformatics journal. 2010;4:5-10
Author contact
![]() Corresponding author: Nouri Neamati. Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, 1985 Zonal Avenue, PSC 304, Los Angeles, CA 90089-9121, USA. Email: neamatiedu. Phone: 323-442-2341. Fax: 323-442-1390.
Corresponding author: Nouri Neamati. Department of Pharmacology and Pharmaceutical Sciences, University of Southern California, School of Pharmacy, 1985 Zonal Avenue, PSC 304, Los Angeles, CA 90089-9121, USA. Email: neamatiedu. Phone: 323-442-2341. Fax: 323-442-1390.