Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Expression of CXCR4 in AML and...
CXCL12/CXCR4 axis mediate the...
The leukemic BM niche
CXCR4 inhibitors-Preclinical...
CXCR4 inhibitors-based therapies...
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2013; 3(1):34-39. doi:10.7150/thno.5150 This issue Cite
Review
Role of CXCR4 in the Pathogenesis of Acute Myeloid Leukemia
Amnon Peled, Sigal Tavor ![]()
Goldyne Savad Institute of Gene Therapy, Hebrew University Hospital, P.O.B 12000, Jerusalem 91120, Israel.
Received 2012-9-2; Accepted 2012-12-10; Published 2013-1-13
Abstract
The Chemokine receptor CXCR4 and its ligand stromal derived factor-1 (SDF-1/CXCL12) are important players involved in cross-talk between leukemia cells and the bone marrow (BM) microenvironment. CXCR4 expression is associated with poor prognosis in AML patients with and without the mutated FLT3 gene.
CXCL12 which is constrictively secreted from the BM stroma and AML cells is critical for the survival and retention of AML cells within the BM. In vitro, CXCR4 antagonists were shown to inhibit the migration of AML cells in response to CXCL12. In addition, such antagonists were shown to inhibit the survival and colony forming potential of AML cells and abrogate the protective effects of stromal cells on chemotherapy-induced apoptosis in AML cells. In vivo, using immune deficient mouse models, CXCR4 antagonists were found to induce the mobilization of AML cells and progenitor cells into the circulation and enhance anti leukemic effects of chemotherapy. The hypothesis that CXCL12/CXCR4 interactions contribute to the resistance of AML cells to signal transduction inhibitor- and chemotherapy-induced apoptosis is currently being tested in a series of Phase I/II studies in humans.
Keywords: CXCR4, CXCL12, AML, Bone marrow, Microenvironment.
Introduction
Acute myeloid leukemia (AML) is a heterogeneous group of diseases characterized by the uncontrolled proliferation of hematopoietic stem cells and progenitors (blasts) with a reduced capacity to differentiate into mature cells [1]. Despite sensitivity to chemotherapy, long-term disease-free survival for AML patients remains low and the majority eventually relapse from minimal residual disease (MRD) [2]. Bone marrow (BM) is the major site for MRD where adhesion of AML cells to bone marrow components may provide protection from the drugs [1]. The chemokine receptor CXCR4 and its ligand stromal derived factor-1 (SDF-1/CXCL12) are important players involved in the cross-talk between leukemia cells and the BM microenvironment [3].
Expression of CXCR4 in AML and its prognostic significance
In 1998, Kanz et al., first published that leukemic blasts (mostly CD34+) from patients with AML expressed variable amounts of CXCR4, which was functionally active, as demonstrated by a positive correlation between the CXCL12-induced migration and the cell surface density of CXCR4 (r = 0.97) [4]. Later, in 2000, the same group published that AML FAB M1/2 blasts did not show calcium fluxes and migration was not stimulated by CXCL12. In myelomonocytic AML (M4/5), however, CXCL12 induced significant calcium fluxes and migration was increased by two-fold. M3 and M4 blasts with eosinophilia (M4eo) showed intermediate activity and M6 blasts showed no functional activity. The capacity of AML cells to respond to CXCL12 by migration and calcium fluxes correlates with CXCR4 cell surface expression levels [5].
Following these publications, in 2002, van Der Schoot, et al., analyzed the CXCL12 dependent migration capacity of cells derived from the BM or peripheral blood (PB) from 26 AML patients [6]. No differences in CXCL12-induced migration or CXCR4 expression were observed between the different AML subtypes. However, more immature leukemic cells expressing CD34, CD38 and HLA-DR were preferentially migrating, whereas cells expressing CD14 and CD36 showed diminished migration. Analysis of paired PB and BM samples indicated that a significantly higher CXCL12-induced migration was observed in AML for CD34(+) BM-derived cells compared to CD34(+) PB-derived cells, suggesting a role for CXCL12 in the anchoring of leukemic cells in the BM. The lower percentage of circulating leukemic blasts in patients with a relatively high level of CXCL12-induced migration also supports this hypothesis [6].
The prognostic significance of CXCR4 expression in patients with AML was examined by different groups. In 2004, Ploemacher, et al., analyzed the expression of CXCR4 together with the expression of CD34 in a series of 90 samples from adult patients with AML [7]. They found that patients with a high CXCR4 expression in the CD34+ subset had a significantly reduced survival and a higher probability of relapse, resulting in a median relapse-free survival (RFS) of only 8.3 months. CXCR4 expression was significantly higher in Fms-like tyrosine kinase-3 (Flt3)/internal tandem duplication (ITD) AML than in Flt3/wild-type (wt) AML. A covariate analysis indicated that the prognostic significance of Flt3/ITDs with respect to RFS was no more apparent when analyzed in conjunction with the expression of CXCR4 in the CD34+ subset, suggesting that the poor prognosis of Flt3/ITD AML might be subordinate to the increased CXCR4 expression. These data suggest that the CXCL12/CXCR4 axis may influence therapy responsiveness and define unfavorable prognosis in AML [7]. Furthermore, in AML with flt3-ITD mutation, the elevated level of PIM1 kinase positively controls cell surface re-expression of CXCR4 from internal pool through phosphorylation of CXCR4 [8].
The prognostic significance of CXCR4 expression in patients with AML who have a normal karyotype and no evidence of FLT3 gene mutations was further examined by Medeiros, et al., in 2007 [9]. In this study, there were 122 AML patients with a median age of 62 years.. CXCR4 was positive in 70 and negative in 52 patients, with complete remission (CR) rates of 58% and 71%, respectively (P =. 09). Multivariate analysis demonstrated that CXCR4 expression, the presence of multilineage dysplasia, and high creatinine levels predicted poorer overall survival (OS) and event-free survival (EFS). The results suggest that CXCR4 expression is associated with poor prognosis also in AML patients with an unmutated FLT3 gene [9].
In 2007, Burger, et al., prospectively evaluated the prognostic implication of CXCR4 in 90 consecutive patients with AML by flow cytometry [10]. Patients could be divided into three groups: those with low CXCR4 expression (n=32), those with intermediate CXCR4 expression (n=26), or those with high CXCR4 expression (n=32). Interestingly, low CXCR4 expression on AML cells correlated with a better prognosis, longer relapse-free and overall survival of 24.3+/-2.9 months for low CXCR4-expressing patients, compared with 17.4+/-3.4 months and 12.8+/-2 months (mean+/-SEM) for patients with intermediate or high expression, respectively. Multivariate analysis revealed CXCR4 expression and unfavorable cytogenetics as independent prognostic factors for disease relapse and survival [10].
In 2009, Guyotat, et al., analyzed, by flow cytometry, the "adhesive" phenotype of AML cells from 36 patients [11]. In a univariate analysis, the main prognostic factor for CR achievement was lower CXCR4 expression (p=0.03). Overall survival (OS) was negatively influenced by higher CXCR4 (p=0.01), very late antigen-4 (VLA-4) (p=0.01), and focal adhesion kinase (FAK) expression (p=0.04). The combination of these markers allowed distinguishing between two prognostic groups: patients overexpressing 2 or 3 factors had a significantly shorter OS (p=0.015). CXCR4, VLA-4 and FAK are new phenotypic markers which could be helpful in establishing risk-stratified therapeutic strategies [11].
In AML, blasts invade the bloodstream and may localize in extramedullar sites, with variations from one patient to another. Herault, et al., hypothesized that a polymorphism in the CXCL12 coding gene (CXCL12 G801A) could influence blast dissemination and tissue infiltration in AML [12]. CXCL12 G801A polymorphism was determined in 86 adult patients and 100 healthy volunteers. 801A carrier status (801G/A, 801A/A) was found to be associated with a higher peripheral blood blast (PBB) count compared with 801G/G homozygous patients (P=0.031) and higher frequency of extramedullar tumor sites (odds ratio 2.92, 95% confidence interval 1.18-7.21, P=0.018). Moreover, the PBB count was correlated with CXCR4 expression (correlation coefficient 0.546, P=0.001) when considering 801A carriers. In conclusion, a polymorphism in the CXCL12 gene was shown to be associated with the clinical presentation of AML and more generally with the risk of distant tissue infiltration by tumor cells [12].
Importantly, the CXCR4 surface expression level may change in physiological conditions like hypoxia. Andreeff, et al., showed that under the condition of reduced partial pressure of oxygen at BM, total and surface CXCR4 expression were upregulated and migration towards CXCL12 was enhanced, suggesting that in the hypoxic area in the BM niche the adhesion and signaling of leukemic cells could be enhanced by increased CXC4 expression [13].
CXCL12/CXCR4 axis mediate the interaction between AML cells and the BM microenvironment
The role of CXCL12/CXCR4 interactions in the control of human AML, cell trafficking and disease progression has been mainly studied using immune deficient mouse models. Most cases of human AML engraft in irradiated non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice [14]. Cases with clinical features of poor prognosis, including FLT3 mutations, tended to engraft efficiently. Nevertheless, AML cells obtained from patients at relapse did not engraft more efficiently than cells obtained from the same patients at initial diagnosis; furthermore, one passage of human AML cells in NOD/SCID mice did not appear to select for increased virulence. Moreover, cDNA microarray analyses indicated that approximately 95% of genes were expressed at similar levels in human AML cells immunopurified after growth in mice, as compared to cells assessed directly from patients [14]. Thus, the growth of human AML cells in NOD/SCID mice could be used to better understand the disease pathogenesis and for the development of novel therapeutics and personalized medicine.
In 2004, Andreeff, et al., evaluated the relationship between engraftment, CXCR4 expression on CD34+ and CD34+CD38- cells, and patient (Pt) clinical/laboratory from 11 Pts [15]. Engraftment, evaluated by Southern blot and CD45 flow cytometric analyses, was observed in murine bone marrow of 6 out of 11 Pt samples, ranging from 0.1% to 73.9% by Southern blot and from 0.1%-36.8% by flow cytometry. No correlation between the level of CXCR4 expression on AML cells and engraftment was observed. Cells with virtually absent CXCR4 expression were able to engraft, and cells from two Pts with high expression levels of CXCR4 did not engraft. Furthermore, anti-CXCR4 antibody failed to block the engraftment of AML cells into NOD/SCID mice [15].
In contrast, in 2004, Lapidot, et al., reported that although some AML cells did not express surface CXCR4, all AML cells tested expressed internal CXCR4 and CXCL12 [16]. Culture of AML cells with CXCL12 promoted their survival, whereas the addition of neutralizing CXCR4 antibodies, CXCL12 antibodies, or AMD3100 significantly decreased it. Pretreatment of primary human AML cells with neutralizing CXCR4 antibodies blocked their homing into the BM and spleen of transplanted NOD/SCID/B2m(null) mice. Furthermore, weekly administrations of antihuman CXCR4 to mice previously engrafted with primary AML cells also led to a dramatic decrease in the levels of human AML cells in the BM, blood, and spleen. Interestingly, the same treatment did not significantly affect the levels of normal human progenitors engrafted into NOD/SCID mice. Taken together, these findings demonstrated the importance of the CXCL12/CXCR4 axis in the regulation of in vivo motility and development of human AML stem cells and identified CXCR4 neutralization as a potential treatment for AML [16].
The leukemic BM niche
Many of the adhesion molecules, chemokine receptors and signaling cascades that are critical to hematopoietic stem cell (HSC) homing, maintenance and egress from the BM niche are shared by the leukemic cells. Leukemic stem (LS) cells home to and engraft within the osteoblast-rich area of the BM, competing for the same microenvironment of normal hematopietic stem cells (HSCs) [17]. Using dynamic in vivo confocal imaging, Lin et al, showed that CXCL12 was expressed in vascular "hot spots" corresponding to the regions that attracted leukemic cells [18]. By directing the leukemic cells in the bone marrow niche, CXCL12 regulate their engraftment and survival [16, 19, 20].
Adhesion of leukemic cells to stromal BM ligands is essential for their survival and proliferation. The adhesion receptor CD44 express on AML cells interact with the BM microenvironment through its major ligand hyaluronic acid which is a component of BM extracellular matrix (ECM). In vivo administration of activating monoclonal anti CD44 to NOD/SCID mice transplanted with human AML markedly reduced leukemic repopulation [21]. In a tetracyclin-inducible mouse model of AML, continuous overexpression of HOXA10 was required to generate AML in primary recipient mice. However, relapsed leukemia was restricted to a population of cells expressing high level of CD44 on the surface and with secondary mutations in additional proto-oncogenes [22]. Another intermediary of the AML cell and ECM interaction is the integrin VLA-4 (very late antigen-) 4, which mediate adhesion to fibronectin and cellular vascular cell adhesion molecule-1 (VCAM1). In a mouse model of MRD, the combined therapy of VLA4 blocking antibodies and chemotherapy prolonged survival [23]. The activation of VLA-4 through CXCL12 stimulation activates adhesion and transendothelial migration of immature CD34+ cells. CXCL12 activates both the integrin VLA-4 and CD44, which are express on AML cells and can enhance their adhesion and chemoresistance [24, 25]. CXCL12 and the interaction of the integrin VLA-4 with the stoma activate multiple pro survival signaling cascades in AML cells. CXCL12 induced phosphorylation of AKT, ERK in AML cells in vitro and CXCR4 antagonists reduced AKT and ERK phosphorylation both in vitro and in vivo in transplanted mice [26, 27].
CXCR4 inhibitors-Preclinical Studies
Antagonists of CXCR4, including AMD3100, induce peripheral mobilization of hematopoietic stem cells and have been approved for clinical use. Several groups explored whether the CXCR4 antagonists affected the trafficking and survival of AML cells in vitro and in vivo.
In 2007, Abboud, et al., demonstrated that AMD3100 can inhibit the CXCL12 dependent transmigration of AML blasts and inhibit outgrowth of leukemia colony forming units [19]. However, AMD3100 did not abrogate stroma-mediated protection from cytarabine-mediated apoptosis, except in the case of one promyelocytic leukemic sample tested; moreover, it did not influence adhesion of blasts to endothelial monolayers. When AML blasts were pretreated with AMD3100, the positive effects of CXCL12 on NOD/SCID engraftment were diminished. This work confirms that AML is influenced by the CXCL12/CXCR4 axis and demonstrates that disruption of this axis by AMD3100 can influence AML [19].
In another study, Rechavi, et al., reported in 2009 that treatment of AML cells with AMD3100 arrested proliferation in AML cell lines and triggered changes that mimicked differentiation, including morphological changes and the expression of myeloid differentiation antigens [20]. In addition, the pan-histon deacetylase inhibitor panbinostat (PS) depleted CXCR4, through proteasomal degradation of CXCR4. PS inhibited the association between CXCR4 and heat shock protein (hsp) 90. Coculture of primary AML cells with PS and AMD3100 synergistically induced apoptosis [28].
CXCL12 signaling plays a key role in the leukemia/bone marrow microenvironment interactions. In 2009, Konopleva, et al., reported that targeting the leukemia microenvironment by CXCR4 inhibition can overcome resistance to kinase inhibitors and chemotherapy in AML [26]. In this study, it was demonstrated that the CXCR4 inhibitor AMD3465 can antagonize CXCL12-induced and stroma-induced chemotaxis and inhibit CXCL12-induced activation of prosurvival signaling pathways in leukemic cells. Further, CXCR4 inhibition partially abrogated the protective effects of stromal cells on chemotherapy-induced apoptosis in AML cells. Notably, CXCR4 inhibition increased the sensitivity of FLT3-mutated leukemic cells to the apoptogenic effects of the FLT3 inhibitor sorafenib. In vivo studies demonstrated that AMD3465, alone or in combination with the granulocyte colony-stimulating factor, induced mobilization of AML cells and progenitor cells into the circulation and enhanced anti-leukemic effects of chemotherapy and sorafenib, resulting in markedly reduced leukemia burden and prolonged survival of the animals [26]. Similarly, in the same year, DiPersio, et al., showed that administration of AMD3100 to leukemic mice induced a 1.6-fold increase in total leukocytes and a 9-fold increase of circulating AML blast counts, which peaked at 3 hours and returned to baseline by 12 hours. Treatment of leukemic mice with chemotherapy plus AMD3100 resulted in decreased tumor burden and improved overall survival compared with mice treated with chemotherapy alone [29].
In 2011, Peled et al., reported that the CXCR4 antagonist, 4F-benzoyl-TN14003 (BKT140) exhibits a CXCR4-dependent preferential cytotoxicity toward malignant cells of hematopoietic origin including AML. In vivo, subcutaneous injections of BKT140 significantly reduced the growth of human AML xenografts [30].
Recently, a comparative study between the CXCR4 antagonists TN140 and AMD3100 suggested that TN140 is more effective than AMD3100 as a monotherapy in AML. TN140 and to a lesser extend AMD3100 induced regression of human CXCR4-expressing AML cells and targeted the NOD/Shi-scid/IL-2Rγnull (NOG) leukemia-initiating cells (LICs) [27]. Another drug in development is the MDX-1338, which is a monoclonal human CXCR4 antibody that inhibited the expansion of AML in xenograft models [31]. Currently a phase I dose escalation study of MDX-1338 with chemotherapy is tested in in relapsed/refractory AML.
CXCR4 inhibitors-based therapies for AML
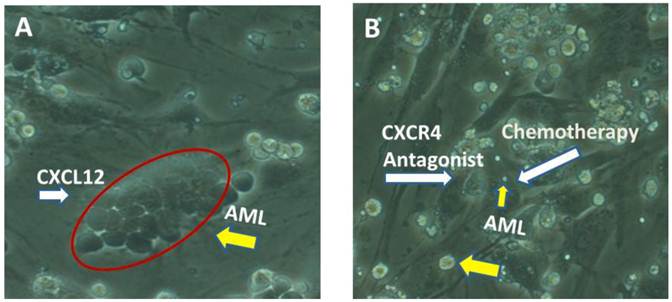
These findings indicate that CXCL12/CXCR4 interactions contribute to the resistance of leukemic cells to signal transduction inhibitor- and chemotherapy-induced apoptosis. Disruption of these interactions with CXCR4 inhibitors represents a novel strategy of sensitizing AML cells by targeting their protective BM microenvironment (Figure 1).
A. AML cells (yellow arrow) are found in close contact with the BM stroma (phase dense cells surrounded by a red line). CXCL12, the ligand of CXCR4, is secreted from the BM stroma and facilitates this interaction which is critical for their retention within the BM. B. Administration of a CXCR4 antagonist will release AML cells (yellow arrow) from the stroma and exposed them to chemotherapy, leading to their cell death (small yellow arrow).
This hypothesis was further tested by DiPersio, et al., in a phase 1/2 study, in which 52 patients with relapsed or refractory AML were treated with the CXCR4 antagonist plerixafor (AMD3100) in combination with mitoxantrone, etoposide, and cytarabine. In the phase 2 part of the study, 46 patients were treated with plerixafor, 0.24 mg/kg/d, in combination with chemotherapy with an overall CR and complete remission with incomplete blood count recovery rate (CRi) of 46%. Correlative studies demonstrated a 2-fold mobilization in leukemic blasts into the peripheral circulation. The authors therefore conclude that the addition of plerixafor to cytotoxic chemotherapy is feasible in AML, and results in encouraging rates of remission with correlative studies demonstrating in vivo evidence of disruption of the CXCR4/CXCL12 axis [32]. The benefit of combing CXCR4 inhibitors with chemotherapy is now being tested in few phase I/II clinical studies in refractory/ relapsed and in newly diagnosed AML patients. Incorporating CXCR4-targeted therapy into AML protocol could not only increase chemosensitivity but also prevent the relapse of the disease by disruption the interaction of residual AML cells with the BM niche.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894-907
2. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A. et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158-65
3. Burger JA, Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. 2009;23:43-52
4. Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor CXCR-4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523-30
5. Mohle R, Schittenhelm M, Failenschmid C, Bautz F, Kratz-Albers K, Serve H. et al. Functional response of leukaemic blasts to stromal cell-derived factor-1 correlates with preferential expression of the chemokine receptor CXCR4 in acute myelomonocytic and lymphoblastic leukaemia. Br J Haematol. 2000;110:563-72
6. Voermans C, van Heese WP, de Jong I, Gerritsen WR, van Der Schoot CE. Migratory behavior of leukemic cells from acute myeloid leukemia patients. Leukemia. 2002;16:650-7
7. Rombouts EJ, Pavic B, Lowenberg B, Ploemacher RE. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104:550-7
8. Konoplev S, Rassidakis GZ, Estey E, Kantarjian H, Liakou CI, Huang X. et al. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Cancer. 2007;109:1152-6
9. Spoo AC, Lubbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. 2007;109:786-91
10. Tavernier-Tardy E, Cornillon J, Campos L, Flandrin P, Duval A, Nadal N. et al. Prognostic value of CXCR4 and FAK expression in acute myelogenous leukemia. Leuk Res. 2009;33:764-8
11. Dommange F, Cartron G, Espanel C, Gallay N, Domenech J, Benboubker L. et al. CXCL12 polymorphism and malignant cell dissemination/tissue infiltration in acute myeloid leukemia. FASEB J. 2006;20:1913-5
12. Fiegl M, Samudio I, Clise-Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2009;113:1504-12
13. Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S. et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med. 2009;206:1957-70
14. Lumkul R, Gorin NC, Malehorn MT, Hoehn GT, Zheng R, Baldwin B. et al. Human AML cells in NOD/SCID mice: engraftment potential and gene expression. Leukemia. 2002;16:1818-26
15. Monaco G, Konopleva M, Munsell M, Leysath C, Wang RY, Jackson CE. et al. Engraftment of acute myeloid leukemia in NOD/SCID mice is independent of CXCR4 and predicts poor patient survival. Stem Cells. 2004;22:188-201
16. Tavor S, Petit I, Porozov S, Avigdor A, Dar A, Leider-Trejo L. et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64:2817-24
17. Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25:1315-21
18. Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435:969-73
19. Liesveld JL, Bechelli J, Rosell K, Lu C, Bridger G, Phillips G 2nd. et al. Effects of AMD3100 on transmigration and survival of acute myelogenous leukemia cells. Leuk Res. 2007;31:1553-63
20. Tavor S, Eisenbach M, Jacob-Hirsch J, Golan T, Petit I, Benzion K. et al. The CXCR4 antagonist AMD3100 impairs survival of human AML cells and induces their differentiation. Leukemia. 2008;22:2151-5158
21. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human myeloid leukemic stem cells. Nat Med. 2006;12:1167-74
22. Quéré R, Andradottir S, Brun AC, Zubarev RA, Karlsson G, Olsson K. et al. High levels of the adhesion molecule CD44 on leukemic cells generate acute myeloid leukemia relapse after withdrawal of the initial transforming event. Leukemia. 2011;25:515-26
23. Matsunaga T, Takemoto N, Sato T, Takimoto R, Tanaka I, Fujimi A. et al. Interaction between leukemic-cell VLA-4 and stomal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9:1158-65
24. Peled A, Kollet O, Ponomaryov T, Petit I, Franitza S, Grabovsky V. et al. The chemokine SDF-1 activates the integrins LFA-1, VLA-4, and VLA-5 on immature human CD34(+) cells: role in transendothelial/stromal migration and engraftment of NOD/SCID mice. Blood. 2000;95:3289-96
25. Avigdor A, Goichberg P, Shivtiel S, Dar A, Peled A, Samira S. et al. CD44 and hyaluronic acid cooperate with SDF-1 in the trafficking of human CD34+ stem/progenitor cells to bone marrow. Blood. 2004;103:2981-9
26. Zeng Z, Shi YX, Samudio IJ, Wang RY, Ling X, Frolova O. et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113:6215-24
27. Zhang Y, Patel S, Abdelouahab H, Wittner M, Willekens C, Shen S. et al. CXCR4 inhinitors selectively eliminate CXCR4-expressing human acute myeloid leukemia cells in NOG mouse model. Cell Death and Dis. 2012
28. Mandawat A, Fiskus W, Buckely KM, Robbins K, Rao R, Balusu R. et al. Pan-histone deacetylase inhibitor panobinostat depletes CXCR4 levels and signaling and exerts synergistic antimyeloid activity in combination with CXCR4 antagonists. Blood. 2010;116:5306-15
29. Nervi B, Ramirez P, Rettig MP, Uy GL, Holt MS, Ritchey JK. et al. Chemosensitization of acute myeloid leukemia (AML) following mobilization by the CXCR4 antagonist AMD3100. Blood. 2009;113:6206-14
30. Beider K, Begin M, Abraham M, Wald H, Weiss ID, Wald O. et al. CXCR4 antagonist 4F-benzoyl-TN14003 inhibits leukemia and multiple myeloma tumor growth. Exp Hematol. 2011;39:282-92
31. Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C. et al. A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows anti tumor activity in vivo. Proceeding of the 100th Annual Meeting of the American Association for Cancer Research; (abstract LB-150). 2009
32. Uy GL, Rettig MP, Motabi IH, McFarland K, Trinkaus KM, Hladnik LM. et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917-24
Author contact
![]() Corresponding author: Amnon Peled, Ph.D. Hadassah Hebrew University Hospital, Gene Therapy Institute, P.O. Box 12000, Jerusalem, 91120 Israel. FAX: 972-2-6430982, Phone: 972-2-677-8780, E-mail: peledorg.il.
Corresponding author: Amnon Peled, Ph.D. Hadassah Hebrew University Hospital, Gene Therapy Institute, P.O. Box 12000, Jerusalem, 91120 Israel. FAX: 972-2-6430982, Phone: 972-2-677-8780, E-mail: peledorg.il.