Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2012; 2(11):1064-1077. doi:10.7150/thno.4181 This issue Cite
Research Paper
Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging
Luis Solorio1, Alexander M. Olear1*, Jesse I. Hamilton2*, Ravi B. Patel1, Ashlei C. Beiswenger1, Jon E. Wallace1, Haoyan Zhou1, Agata A. Exner1 ![]()
1. Department of Biomedical Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH 44106.
2. Department of Biomedical Engineering, Johns Hopkins University, 3400 North Charles St., Baltimore, MD 21218.
* Authors contributed equally to this work.
Received 2012-2-1; Accepted 2012-3-19; Published 2012-11-8
Citation:
Solorio L, Olear AM, Hamilton JI, Patel RB, Beiswenger AC, Wallace JE, Zhou H, Exner AA. Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging. Theranostics 2012; 2(11):1064-1077. doi:10.7150/thno.4181. https://www.thno.org/v02p1064.htm
Other stylesAbstract
In situ forming implants (ISFIs) have shown promise in drug delivery applications due to their simple manufacturing and minimally invasive administration. Precise, reproducible control of drug release from ISFIs is essential to their successful clinical application. This study investigated the effect of varying the molar ratio of different molecular weight (Mw) poly(D,L-lactic-co-glycolic acid) (PLGA) polymers within a single implant on the release of a small Mw mock drug (sodium fluorescein) both in vitro and in vivo. Implants were formulated by dissolving three different PLGA Mw (15, 29, and 53kDa), as well as three 1:1 molar ratio combinations of each PLGA Mw in 1-methyl-2-pyrrolidinone (NMP) with the mock drug fluorescein. Since implant morphology and microstructure during ISFI formation and degradation is a crucial determinant of implant performance, and the rate of phase inversion has been shown to have an effect on the implant microstructure, diagnostic ultrasound was used to noninvasively quantify the extent of phase inversion and swelling behavior in both environments. Implant erosion, degradation, as well as the in vitro and in vivo release profiles were also measured using standard techniques. A non-linear mathematical model was used to correlate the drug release behavior with polymer phase inversion, with all formulations yielding an R2 value greater than 0.95. Ultrasound was also used to create a 3D image reconstruction of an implant over a 12 day span. In this study, swelling and phase inversion were shown to be inversely related to the polymer Mw with 53kDa polymer implants increasing at an average rate of 9.4%/day compared with 18.6%/day in the case of the 15 kDa PLGA. Additionally the onset of erosion, complete phase inversion, and degradation facilitated release required 9 d for 53 kDa implants, while these same processes began 3 d after injection into PBS with the 15 kDa implants. It was also observed that PLGA blends generally had intermediate properties when compared to pure polymer formulations. However, release profiles from the blend formulations were governed by a more complex set of processes and were not simply averages of release profiles from the pure polymers preparations. This study demonstrated that implant properties such as phase inversion, swelling and drug release could be tailored to by altering the molar ratio of the polymers used in the depot formulation.
Keywords: in situ forming implants, polymer molecular weight, phase inversion, biodegradable polymers, controlled release, ultrasound, drug delivery, PLGA.
Introduction
Encapsulation of therapeutic agents within biocompatible polymeric matrixes is often a necessity for parenteral administration. This type of delivery is of particular interest for drugs that are rapidly metabolized or hydrolyzed, have a poor oral availability, or have a narrow therapeutic index [1-3]. Depending on the application, dissolution of the drug from polymer delivery systems can occur over periods of days to years leading to improved patient compliance as well as elevated local concentrations of drug with reduced systemic involvement [1-5]. Numerous drug encapsulation strategies have been implemented ranging from particulate systems to solid implants. Among these, injectable, in situ forming implants (ISFI) are especially compelling for local delivery applications because they do not require invasive placement, and their composition is easily modified and safe for heat sensitive drugs and biologics.
One example of this system is an injectable phase sensitive formulation, developed by Dunn et al, which has been used to effectively treat prostate cancer and periodontal disease clinically [6-11]. This system consists of a biodegradable hydrophobic polymer (typically PLGA), dissolved in a biocompatible organic solvent which can be mixed with an active drug. Once the polymer solution contacts the aqueous environment, counter exchange of solvent and water begins, resulting in the precipitation of the polymer from the unstable tertiary system leading to the formation of a solid drug eluting depot through a process known as phase inversion [6, 8, 12-16]. Since the implants are initially a liquid solution, drug loading is performed simply through the addition of a powder form of the drug into the polymer/solvent mixture [5, 10, 11, 13, 17-21]. Additionally, the solution can be injected through a minimally invasive procedure using image guidance [1, 5, 22]. Due to the inherent versatility and ease of manufacturing of these implants, a number of different applications have been investigated ranging from the intratumoral delivery of drug in order to treat solid tumors to a tissue engineering scaffold designed to release DNA [9, 23-27]. Furthermore, treatment can be halted by surgically removing the implant if the need arises [10, 11].
Many parameters have been shown to affect the drug release profile of injectable implants including solvent polarity, polymer concentration, and polymer type [4, 11-13, 19, 28-42]. Through the use of poorly miscible solvents which lead to a slowly formed, dense polymer matrix, implants with a near zero order release have been developed [13, 34, 43, 44]. Despite the optimal release profile, their use has been somewhat limited clinically due to their high viscosity which limits the injectability of the polymer solution [35, 41, 44]. Conversely, fast phase inverting systems have optimal viscosity, but have been shown to release a large burst of drug due to formation of a heterogeneous polymer matrix with a highly interconnected porous network [13, 19-21, 26, 45]. In order to limit the burst release, excipients have been used [21, 31, 35]. These additives reduce burst by decreasing the diffusivity coefficient of drug through the porous network within the depot [13, 21, 35]. While they have shown to be effective in vitro, there efficacy is somewhat limited in vivo [26].
Varying the polymer molecular weight (Mw) is an alternative method by which drug release and phase inversion can be modified for implants formed both in vitro and in vivo [11, 13, 21, 26, 46]. Previous research has demonstrated that the average Mw of PLGA influences both the release profile and implant morphology [21, 26, 30, 39, 40, 47], with lower Mw implants having greater overall release and phase inversion than the higher Mw counterparts [30, 39, 40, 47, 48]. These differences have been attributed to the higher hydrophilicity and degradation rate of the lower Mw polymers [13, 19, 30, 39, 40, 47]. For many therapeutic applications, the ideal release profile for such an agent would have an intermediate release than what is obtained from the pure polymer types. The aim of this study was to demonstrate that using PLGA Mw blends could be a simple way to optimize the release of a small-molecule, hydrophilic drug without the use of additional excipients. Blends of different Mw PLGA polymers were investigated both in vivo and in vitro. Effect of the blends on the rate of implant formation, evolution of implant morphology, polymer degradation and erosion were investigated using a noninvasive imaging technique developed previously by our group [22, 26]. Briefly, diagnostic ultrasound was used to visualize and quantify phase inversion in real-time. As the liquid implant formulation begins to solidify upon contact with water, the acoustic impedance of the polymer solution changes, and a higher proportion of incoming ultrasound waves are backscattered. These changes in acoustic impedance can be used to visualize the rate of phase inversion, providing insight into changes in implant morphology both in vitro and in vivo. This is of interest since phase inversion of ISFIs has been thought to govern much of the early drug release properties [13, 19, 45, 49], yet the undisrupted behavior of ISFI has been difficult to quantify. In this study we show a direct relationship between specific morphological parameters and alterations in degradation, erosion and subsequent drug release profiles resulting from PLGA Mw variations.
Materials and Methods
Materials
All materials were used in the state obtained from the respective companies. Poly (DL-lactide-co-glycolide) (PLGA 50:50 2A, MW 15kDa, inherent viscosity 0.16dl/g; 3A, MW 29kDa, inherent viscosity 0.28 dl/g; 4A, MW 53kDa, inherent viscosity 0.46 dl/g) was obtained from Lakeshore Biomaterials, Birmingham, AL. N-methyl pyrrolidinone (NMP) and sodium fluorescein (MW 376.28) were obtained from Sigma Aldrich, St. Louis, MO. Agarose was acquired from Fischer Scientific, Waltham, MA.
Preparation of Polymer Solutions
Fluorescein was used as a mock low Mw mock hydrophilic drug. Polymer solution was prepared using a mass ratio of 39:60:1 PLGA to NMP to fluorescein. First, fluorescein was dissolved in NMP for 1 hr on an incubated orbital shaker (37ºC, 90 RPM). Next, PLGA was added to the NMP:fluorescein solution and allowed to dissolve overnight in the orbital shaker protected from light. Blend formulations of 1:1 molar ratios of the three different Mw polymers were produced for investigation (15kDa:29kDa, 15kDa:53kDa, 29kDa:53kDa). Additionally, formulations consisting of pure non-blended polymer were produced (15kDa, 29kDa, and 53kDa).
Ultrasound Characterization
Implants were imaged using ultrasound as previously described [22, 50]. Briefly, an agarose phantom containing a 1 ml void was filled with phosphate buffered saline (PBS, pH 7.4). Then polymer solution (44.3±3.6 mg) was injected into the PBS. Images were taken immediately after injection into PBS, and then after 0.5, 1, 2, 4, 6, and 8 h during the first day (d), then daily for 10 d. Images were obtained using a 12 MHz transducer (Aplio XG, Toshiba Medical Systems). Implants were kept at 37°C on an incubated orbital shaker in 10 ml of warm PBS for the duration of the study. Image analysis was performed using a custom MatLab code (MathWorks Inc., Natick, MA) which selected the implant shell, applied a threshold (determined through a parametric segmentation of mixed Gaussians) to the entire image to create a binary image, and summed the pixel values to determine the area of the formed polymer shell and the total cross-sectional area of the implant [22, 26, 50]. The rate of phase inversion was quantified using the ratio of the polymer shell area relative to the total cross-sectional area, and the change in cross-sectional area over time was used to monitor implant swelling [22, 26, 50]. Linear regions in the swelling profile were determined by evaluating derivatives of the swelling plots. A 3-D ultrasound tomography analysis was performed by embedding implants in a 1% agarose phantom. The phantom was then placed on a MSL series linear stage (Newmark systems, MSL-25-11), with the US transducer fixed above the actuated stage. Images were taken every 200 µm through the volume of the implant. The 2-D ultrasound data was then reconstructed into a 3-D volume using AMIRA (Visage Imaging GmbH) (Supplemental data).
Drug Release
50 µl of implant solution (44.3±3.6 mg) was injected into a 10 ml 37ºC PBS bath (pH 7.4). Implant mass was recorded immediately, and then 1 ml of the sample solution was removed and then replaced by 1 ml of warm fresh PBS in order to maintain sink conditions. Samples were taken on the first day at 0, 0.5, 1, 2, 4, 6, and 8 h, then once daily for 14 d, and on days 16, 18, and 21. After 21 d, the implants were removed and added to 5mL of 2M NaOH overnight in order to determine the mass of any residual drug. Fluorescence was measured using a Tecan Ltd, Infinite 200 Series plate reader, with excitation wavelength 485nm and emission wavelength 525nm. The concentration of fluorescein was measured by comparison to known established standard concentrations, and the cumulative drug release was then calculated from the obtained fluorescence data. The diffusion and degradation phases of release were determined by evaluating derivatives of the release plots. As previously described, an empirical relationship was developed to describe how drug release and phase inversion were correlated using a non-linear mathematical fit to F(x)=S0(1-e(-τx))+mx [50]. F(x) is the percent phase inversion, x represents the percent drug release (%), S0 represents the initial polymer precipitation (%), τ is the time delay (1 /%release) and m is the proportionality constant that relates phase inversion to drug release (% / %release).
Erosion and Degradation
Implant erosion was determined by monitoring the change of implant mass with respect to time. After implant formation, the initial mass was recorded. 1 ml of bath-solution was discarded daily and replaced with 1 mL of fresh PBS solution to maintain sink conditions. At each respective time point (every other day for 21 d) the implant was removed from solution, frozen, then lyophilized for 5 d, and the final implant mass was recorded. The degradation was monitored by evaluating changes in the weight average Mw over time as previously described [50]. The implants were prepared for GPC analysis by first dissolving the lyophilized implants in tetrahydrofuran (THF) and then filtered using a 0.45µm syringe filter. Mw was determined relative to narrow polystyrene standards using an Agilent 1200 series liquid chromatography system, a refractive index and variable wavelength detector, and two American Polymer Standards linear bed GPC columns (Mentor, OH) at a flow rate of 1 ml/min.
Figure 1
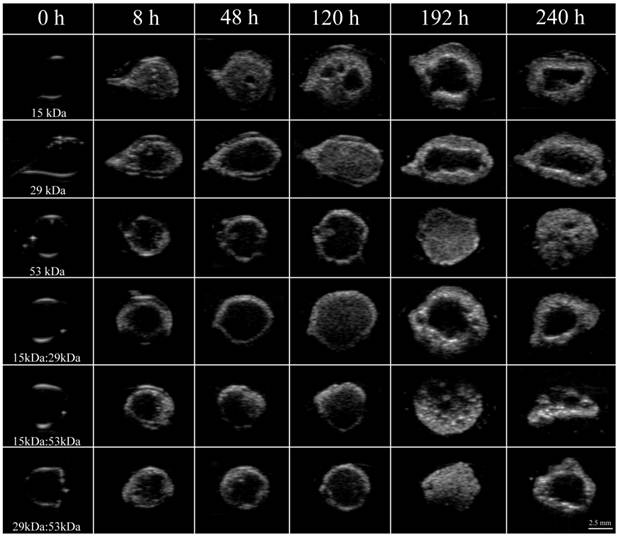
Representative ultrasound images of the same implant over a period of 10 days for all six formulations examined in this study. Whiter areas represent precipitated polymer while darker areas suggest a mixture of water, solvent and remaining dissolved PLGA. Row 1 is 15 kDa implants, row 2 is 29 kDa implants, row 3 is 53 kDa implants, row 4 is 15kDa:29kDa blend, row 5 is 15kDa:53kDa blend, and row 6 is 29kDa:53kDa blend. Pore is indicated by the arrow.
In Vivo Analysis
All animal studies were performed as previously described following protocols approved by the Case Western Reserve University Institutional Animal Care and Use Committee [22, 26]. Briefly, five 24-week old male Sprague-Dawley rats with an average body weight of 565±58 g (Charles River Laboratories Inc., Wilmington, MA) were anesthetized using 1% isoflurane with an oxygen flow rate of 1 l/min (EZ150 Isoflurane Vaporizer, EZ AnesthesiasTM). Implant solution (50 to 70 µl) was injected under the dorsal skinflap in five locations using an 18-gauge hypodermic needle. The depots were then imaged using ultrasound with a 12 MHz transducer 1 h, 4 h, 8 h, 24 h, and 48 h after implantation. Cumulative release was determined by subtracting the residual drug in the implant from theoretical drug loading. Residual drug content was determined by first dissecting out the implants after euthanasia, then degrading the implants in 5 ml of 2 M NaOH overnight. The mass of fluorescein was determined by comparison to a standard curve using a Tecan 200 series plate reader at an excitation wavelength of 485 nm and an emission wavelength of 525 nm [22, 26].
Statistical Analysis
Statistical significance was determined using ANOVA (p<0.05, N=4 for in vitro studies and N=5 for in vivo studies). Significant difference among groups was established using a Tukey multiple comparison test, with analysis performed using Minitab. Unless otherwise noted, data is reported as mean ± standard deviation.
Results
Ultrasound Imaging
Immediately after injection of the implant solution into PBS, a thin polymer shell could be visualized in all implants as a result of mass transfer events increasing the acoustic impedance mismatch of the polymer and the surrounding PBS (Figure 1). After the initial shell formation, the implants continued to transition into solid depots, which resulted in an increase in ultrasound backscatter within the interior of the implants (Figure 1). After phase inversion had completed, a pore formed in the center of the implants that continued to expand over the duration of the experiment. The time required for a pore to develop was inversely related to the polymer Mw (Figure 1).
Phase Inversion
In all cases, the implants rapidly precipitated reaching a period of limited change within the first 8 h of exposure to the bath solution. Formulations using either 15 kDa or 29 kDa PLGA achieved a pseudo-steady state after 4 h in the bath-side solution (precipitating at 77.8±1.9% and 56.0±1.5% of the total cross-sectional area respectively), with implants formulated using 15 kDa PLGA precipitating significantly more than all other formulations. Implants using the 53 kDa PLGA, 15 kDa:29 kDa, or 15 kDa:53 kDa polymers all reached a plateau after 6 h in PBS with the precipitated polymer occupying 53.7±3.3%, 50.2±3.4%, and 62.2±3.1% of the cross-sectional area respectively. Finally, depots consisting of 29 kDa:53 kDa PLGA reached a plateau of 63.7±5.3% after 8 h in the bath-side solution (Figure 2).
After the initial rapid period of phase inversion, final polymer precipitation varied with the polymer used, with maximum polymer precipitation taking as little as 3 d or as long as 9 d. Implants containing 15 kDa PLGA underwent faster phase inversion than implants without the low Mw polymer. Implants formulated using only 15 kDa PLGA completed phase inversion the fastest, reaching a maximum precipitated area of 84.5±4.0% within 3 days.
Figure 2
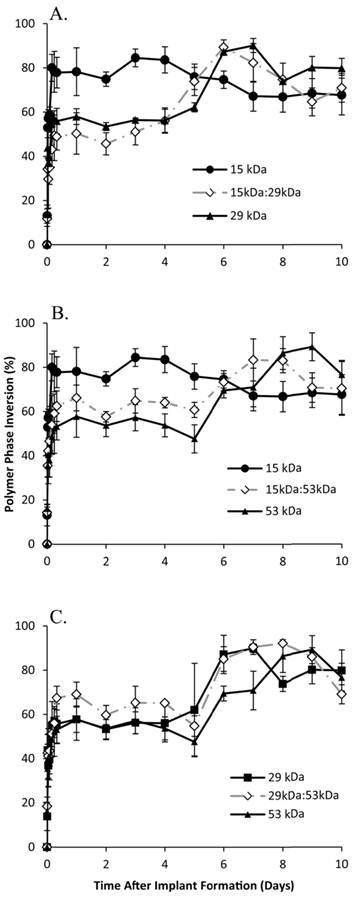
Quantitative formation data of ISFIs over 10 days, with the 15 kDa, 29 kDa, and 15kDa:29kDa blend (A); 15 kDa, 53 kDa, and 15kDa:53kDa blend (B); 29 kDa, 53 kDa, and 29kDa:53kDa blend (C). Data for the PLGA blends were plotted along with the corresponding pure PLGA to demonstrate intermediate behavior of the implants.
The mixture of 15 kDa PLGA with 29 kDa PLGA increased the amount of time for phase inversion, reaching a maximum of 89.3±1.7% after 6 d. Both implants formulated using the 15 kDa:53 kDa blend and with 29 kDa PLGA alone completed phase inversion after 7 days, reaching a maximum of 83.4±9.5% and 90.1±2.0% respectively (Figure 2).
The blend of 29kDa:53kDa PLGA required 8 d to completely phase invert, with the precipitated polymer occupying 92.2±0.6% of the cross-sectional. Finally, the implants formulated using only 53 kDa PLGA completely phase inverted after 9 d in PBS, reaching the maximum precipitation (89.3±6.3%) (Figure 2).
Swelling
The 15 kDa implants initially increased in size, expanding to 1.26±0.16 times the original cross-sectional area within 30 m in solution, with negligible changes occurring within the first 24 h after the initial expansion. Between 24 and 48 h in buffer, implants significantly increased in size, increasing to 1.74±0.15 times the initial cross-sectional area, expanding by 47.3% within that 24 h period. Between 48 and 120 h, implants continued to swell an average rate of 18.6% per day. After 120 h in buffer, the implants showed no significant changes in cross-sectional area, remaining at 2.36±0.04 times the initial area, for the duration of the study (Figure 3).
Implants formulated using the 29 kDa PLGA achieved a pseudo-steady state 2 h after injection into the bath-side solution, increasing 1.19±0.01 times larger than the original cross-sectional area. After 24 h in solution, the implants increased an average of 10.2% per day (from 1.20 to 1.81 times the original area). After 192 h the implants showed a rapid decrease in cross-sectional area, shrinking 58.7% over the final 48 h of the study (Figure 3). A 3-D reconstruction of an implant formulated using the 29 kDa polymer demonstrates the expansion (1 d, 3 d, and 10 d) and collapse (12 d) of the drug eluting depot over the course of 12 d (Figure 4). Implants formulated using the 15kDa:29kDa blend achieved an initial plateau after 4 h in buffer. After 24 h, the cross-sectional area of depots formulated using the 15kDa:29kDa polymer blend increased at a rate of 14.8% per day, but began to decrease in size after 192 h in the bath-side solution (Figure 3).
All implant formulations using the 53 kDa polymer, initially decreased in size. Depots formulated using only the 53 kDa PLGA began to approach the original area after 24 h in solution (0.98±0.04 times the initial area), while the 15 kDa:53 kDa implants required 48 h and the 29 kDa:53 kDa PLGA depots approached the original cross-sectional area after 2 h in solution. Implants formulated using the 53 kDa polymer expanded an average of 9.4% per day after 24 h in solution, and began to decrease in size after 192 h (Figure 3). Between 48 h and 168 h, depots formulated using the blend of 15 kDa:29 kDa PLGA began to increase at a rate of 5.5% per day. While the implants formulated using the 29 kDa:53 kDa PLGA blend increased at a rate of 7.4% per day after 8 h in solution for the duration of the study. Results are summarized in Table 1.
Figure 3
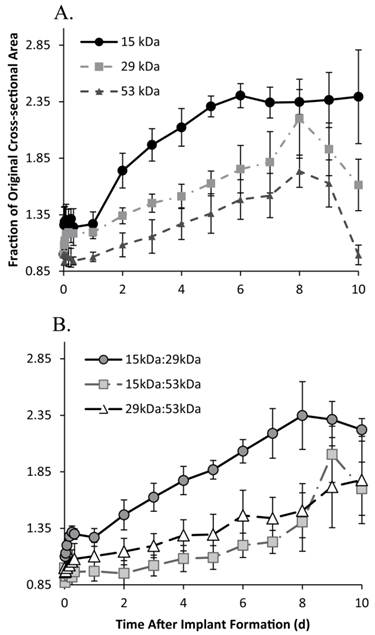
Change in implant cross-sectional area over time, for the 15 kDa, 29 kDa, and 53 kDa implants (A); and 15kDa:29kDa blend, 15kDa:53kDa blend, and 29kDa:53kDa blend (B).
Table 1
Parameters characterized using ultrasound imaging.
| Ultrasound Characterization | 15kDa | 29kDa | 53kDa | 15kDa:29kDa | 15kDa:53kDa | 29kDa:53kDa |
|---|---|---|---|---|---|---|
| Maximum Phase Inversion (d) | 3 | 7 | 9 | 6 | 7 | 8 |
| Maximum Phase Inversion (%) | 84.5±4.0 | 90.1±2.0 | 89.3±6.3 | 89.3±1.7 | 83.4±9.5 | 92.2±0.6 |
| Rate of Swelling (%/d) | 18.6 | 10.2 | 9.4 | 14.8 | 5.5 | 7.4 |
Figure 4
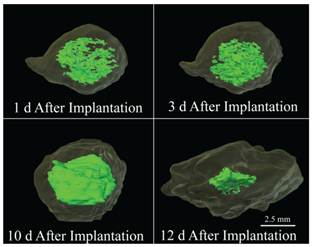
3-D reconstruction of a 29 kDa implant over the course of 12 days, the light green region is the hyperechoic domain (precipitated polymer) and the interior bright green region is the hypoechoic domain indicating a homogenous liquid region.
Drug Release
Phase sensitive ISFIs have release profiles that typically consist of three phases: burst, diffusion, and degradation. During the initial burst period of release, which occurs over the course of the first 24 h, implants containing the 53 kDa polymer had the statistically highest burst release of mock drug relative to the implants formulated with only one polymer type (Figure 5). The 29kDa:53kDa blend had the highest overall burst, followed by implants with only the 53 kDa PLGA, and then the 15kDa:53kDa (burst release of 31.0±3.0%, 30.6±2.3%, and 29.6±2.1% respectively). Implants formulated with using only the 29 kDa polymer averaged a lower burst release than the other implant formulations, followed by the 15 kDa and the 15kDa:29kDa formulations (22.7±2.9%, 24.8±1.7%, and 27.1±3.2% respectively).
The duration of diffusion and degradation phases was dependent on the Mw of the polymer. The transition between phases was determined by evaluating the derivative of release. The numerical derivative doubled for all formulations except for implants formulated using only the 15 kDa PLGA. Implants fabricated using the 29 kDa polymer alone, began to show elevated release after 7 d in the bath-side solution; this transition occurred after 9 d for implants formulated using the 53 kDa polymer. Blend formulations that contained the 15 kDa polymer transitioned into the degradation phase after 6 d in PBS, but the 29kDa:53kDa blend took 8 d (Figure 5). While implants formulated using the 15 kDa polymer did not have distinct degradation and diffusion phases, the rate of drug dissolution per day was statistically greater for the 15 kDa polymer than any of the other implant formulations over the entire time course of the study (5.2±0.4 %/d). Among the implant formulations with distinguishable phases of release, those formulated with the 29 kDa polymer, the 15kDa:29kDa blend, and the 29kDa:53kDa blend had a statistically greater rate of dissolution than implants using the 53 kDa polymer and the 15kDa:53kDa blend (Table 2). During degradation facilitated release, all implant formulations with distinguishable release phases had statistically equivalent release kinetics (Table 2). The release characteristics are summarized in Table 2.
Table 2
Average release of fluorescein at each release phase.
| Release | 15 kDa | 29 kDa | 53 kDa | 15kDa:29kDa | 15kDa:53kDa | 29kDa:53kDa |
|---|---|---|---|---|---|---|
| Burst (%) | 24.8±1.7ϯ | 22.7±2.9 | 30.6±2.3* | 27.1±3.2 | 29.6±2.1 | 31.0±3.0 |
| Diffusion (%/d) | 5.2±0.4**,£ | 2.6±0.7** | 1.2±0.6** | 2.2±0.6 | 1.8±0.4 | 2.0±0.3 |
| Degradation (%/d) | - | 4.14±0.4 | 3.2±0.6 | 4.1±0.3 | 3.5±1.0 | 4.4±0.7 |
| Lock Down (%) | 83.6±3.1+ | 76.9±6.4 | 70.5±5.6+,± | 79.9±6.2 | 76.9±6.7 | 81.4±5.0± |
ϯ indicates that the 15 kDa polymer had a statistically different burst than the 29kDa:53kDa PLGA blend.
*indicates that the 53 kDa blend had statistically greater burst than the 15 kDa or 29 kDa blend, but equivalent burst to the blends.
**indicates that the pure polymer types had statistically different diffusion release from each other.
£ indicates statistically different diffusion release from the blends.
+ indicates a statistical difference between 15 kDa and 53 kDa implants.
± indicates the statistical difference between the 53 kDa and 29kDa:53kDa blend.
Figure 5
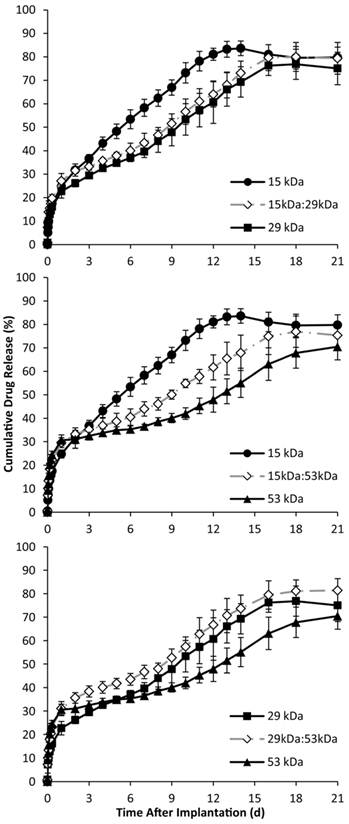
Cumulative in vitro release of fluorescein over time, with the 15 kDa, 29 kDa, and 15kDa:29kDa blend (A); 15 kDa, 53 kDa, and 15kDa:53kDa blend (B); 29 kDa, 53 kDa, and 29kDa:53kDa blend (C). Blends were plotted with corresponding pure PLGA to demonstrate intermediate behavior of the implants.
Correlation of Drug Release with Phase Inversion
The percent of drug released was correlated with phase inversion during the burst phase of release (Figure 6). The S0 parameter was used to describe the initial echogenicity of the implants, with the 15 kDa polymer implants having the highest value, followed in order of decreasing echogenicity by the 29 kDa polymer, the 29kDa:53kDa polymer, the 15kDa:53kDa polymer, the 53kDa polymer, and the 15kDa:29kDa blend (Table 3). The transition to the linear region, τ, was fastest with implants formulated using the 53 kDa polymer and the associated blends (table 3). The 15kDa:29kDa implants transitioned more rapidly than implants formulated using either the pure 15 kDa polymer or 29 kDa PLGA (Table 3). All implant formulations except for those formulated using only the 15 kDa polymer had a sensitivity greater than 1 within the linear range.
Figure 6
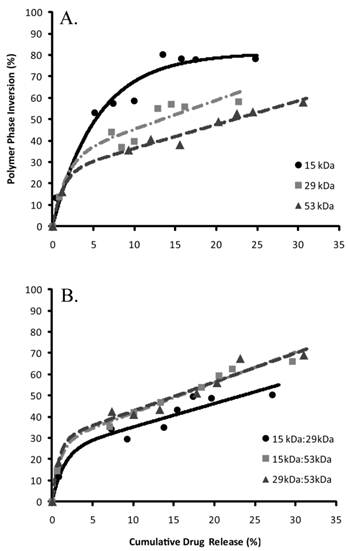
Correlation of phase inversion relative to drug release, (A) pure polymer types and (B) are the polymer blends.
Erosion and Degradation
A rapid initial decrease in mass occurred for all implant formulations, with an average mass loss of 53±1.3% within 24 h after injection into the bath-side solution. Implants formulated with 15 kDa PLGA had the highest rate of erosion, reaching a residual mass of 38.0±2.1% within 5 d in buffer (Figure 7). Erosion was a slower process for all other polymer formulations, requiring 7 d to achieve a residual mass lower than 40% for the 15kDa:29kDa polymer implants (35.3±1.0%), 8 d for the 29 kDa implants (37.6±2.8%), 10 d for both the 15kDa:53kDa blend and the 29kDa:53kDa blend (37.7±2.2% and 37.2±1.2%), and 12 d for the 53 kDa implants (35.0±0.5%). Polymer degradation was faster in larger Mw polymers and decreased with the initial Mw (Figure 8).
Figure 7
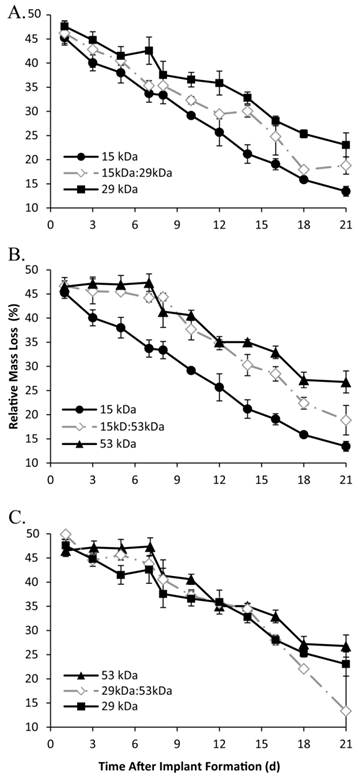
Implant erosion over time, with the 15 kDa, 29 kDa, and 15kDa:29kDa blend (A); 15 kDa, 53 kDa, and 15kDa:53kDa blend (B); 29 kDa, 53 kDa, and 29kDa:53kDa blend (C).
In Vivo Formation and Drug Release
Polymer precipitation occurred rapidly, reaching a plateau within the first 4 h of implantation, with negligible changes occurring for the remainder of the study (Figure 9). The average gray-scale value was significantly lower in vitro than in vivo over all time points, and followed a similar trend as phase inversion (Figure 10). The release of fluorescein was significantly higher in vivo than in vitro releasing 25.3±2.6% of the mock drug released within the first hour after implantation, increasing to 42.0±4.5% after 4 h, with significantly less release occurring during the following 4 h. A secondary burst of drug was released between 8 h and 24 h after implantation, with negligible changes occurring for the duration of the study. Correlation of the burst release and phase inversion resulted in an initially linear relationship, with a sensitivity greater than 1 (Table 3).
Figure 8
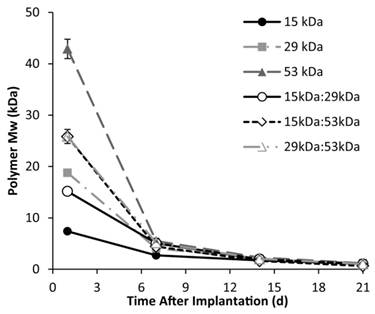
change in PLGA Mw over 21 days in response to hydrolysis of the polymer.
Table 3
Parameters evaluated for mathematical fit of phase inversion and drug release.
| Variables | 15kDa | 29kDa | 53kDa | 15kDa:29kDa | 15kDa:53kDa | 29kDa:53kDa | In Vivo |
|---|---|---|---|---|---|---|---|
| S0 (% phase inversion ) | 79.4 | 31.9 | 25.3 | 24.1 | 27.6 | 29 | 1.0 |
| τ (1/% drug release) | 0.2 | 0.6 | 0.8 | 0.7 | 1.0 | 1.2 | -0.1 |
| m (% phase inversion/% drug release) | 0.1 | 1.3 | 1.1 | 1.1 | 1.4 | 1.4 | 1.6 |
| R2 | 0.97 | .95 | .99 | .96 | .99 | .98 | .99 |
Figure 9
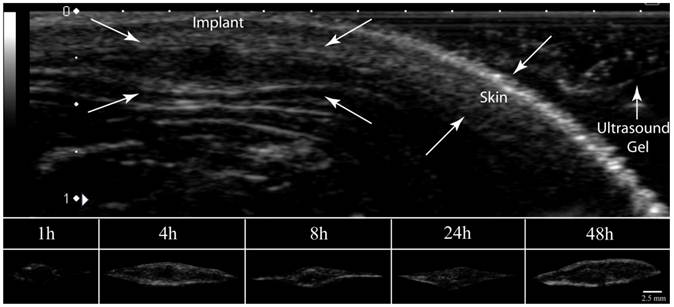
Representative ultrasound image of implants formed subcutaneously in a rat model. The arrows indicate the implant, skin, and ultrasound gel.
Figure 10
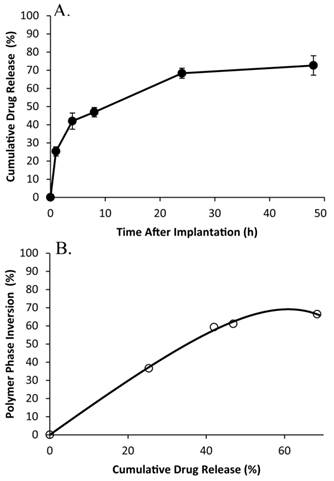
Cumulative release of fluorescein from the 15kDa:29kDa implants in vivo (A), and the correlation of phase inversion to drug release (B).
Discussion
ISFI systems provide a minimally invasive platform for the sustained local delivery of drugs, while concurrently improving patient compliance and minimizing fabrication costs. Due to the inherent flexibility of this implant system, any number of therapeutic agents can be administered and loading can easily be customized. In order to better tailor the implant system for the desired application, it is important to understand the role of implant composition, particularly the Mw of the polymer used in the ISFI. The purpose of this study was to evaluate the effect of blending various Mw polymers to develop implants with customizable intermediate properties.
Since ISFIs can be characterized noninvasively with ultrasound, the transition of ISFIs from a liquid solution to a solid drug eluting depot could be observed longitudinally in a single implant. All implants initially formed a polymer shell immediately after contact with an aqueous environment, followed by a more gradual transition within the interior of the depot. In all cases, the implants formed a pore within the center of the depot that increased in diameter over time (Figure 1). The formation of macrovoids has been reported in asymmetric membranes manufactured using phase inversion with solvents that are highly miscible with the nonsolvent [14, 16, 51-53]. In the case of asymmetric membrane formation, the phase inversion occurs in two steps [16]. First, a dense polymer shell forms that creates a barrier for diffusion, then the rate of phase invasion slows down leading to the development of counter diffusion gradients between the solvent and nonsolvent [16]. This process leads to the development of macropores within these membranes as the less stable solvent-rich interior separates into polymer lean domains and more stable polymer rich regions [16]. In the case of ISFIs using NMP (a highly water miscible solvent), we can see the rapid formation of the shell, followed by the slower development of echogenic signal within the interior of the implant. Therefore, the central pore observed may be the formation of a macrovoid within the center of the implant at the location where the solvent concentration is the highest.
A consequence of the polymer shell and the implant size is the potential for development of a pH gradient in addition to the solvent gradient. It has been reported that when implants are large and fabricated using bulk eroding polymers, the interior of the implants degrades more rapidly than the portion exposed to the bath-side solution [54-56]. Since the polymer shell is in constant contact with the bath-side, the newly formed acidic oligomers from the degrading polymer shell are rapidly lost to the surrounding solution [54, 56]. Conversely, the acidic products formed within the interior, are not readily released to the exterior solution due to the low diffusivity of the polymer shell. This difference in oligomer retention results in the development of a pH gradient that leads to the autocatalysis of the polymer within the implant interior and an increase in implant osmolarity [54-57]. The presence of these degradation products would ultimately lead to the complete solidification of the phase inverting implant by increasing the diffusion of water into the interior, which would stabilize the inner most domains. While the initial pore formation may be a result of a solvent gradient, the dynamic nature of the central pore is most likely a function of the polymer autocatalysis. This process was observed on the 2- and 3-D ultrasound images of the implant. Here, initially the interior of the implant was hyperechoic which we hypothesize to be the polymer lean droplets consisting of primarily solvent and nonsolvent (Figure 4, 1 d). With time, these nucleation sites (of polymer lean regions) aggregate (Figure 4, 3 d) and form a more homogenous gel like interior, with nonsolvent concentrations below the critical water concentration. After the implant completely phase inverts the central pore consisting of degraded oligomers and nonsolvent could be observed as a central hypoechoic domain, leading to an elevated implant volume (Figure 4, 10 d). The implant then collapses from a spherical implant structure into a flat disk structure, which may be a result of continued degradation leading to the loss of mechanical properties in the outer polymer shell (Figure 4, 12 d).
As with the formation of the central pore, phase inversion was also a function of the polymer Mw. The effect of polymer Mw on phase inversion has been hypothesized to be a result of implant osmolarity, polymer hydrophobicity, affinity for solvent, and a decreased diffusivity of the outer shell [13, 22, 58]. Therefore, implants formulated using lower Mw PLGA phase invert more rapidly than implants formulated with higher Mw polymers. As a consequence of the relationship of Mw to phase inversion, implants formulated using blends had phase inversion profiles that were intermediate to the pure polymer types (Figure 2). Additionally, polymer swelling was inversely related to the polymer Mw, with the blends having intermediate swelling profiles relative to the pure polymer types. The only exception being the 15kDa:53kDa blend, which had a swelling profile comparable to the 53 kDa PLGA implants (Figure 3). This difference in swelling behavior could be a result of residual solvent concentration within the polymer changing the initial implant osmolarity, a result of shell diffusivity, or a consequence of polymer hydrophobicity.
When the implant erosion was evaluated, there was an initial rapid release of solvent leading to an elevated mass loss during the first 24h for all polymer formulations. The erosion profile is in stark contrast to what is observed from preformed polymer implants where only negligible changes in the implant mass occur initially followed by a period of rapid erosion. After initial solvent loss, a similar process is observed with ISFIs, in that the onset of erosion (residual mass less than 39% of the initial mass) is delayed. The lag time for erosion increased with Mw, with the blends having intermediate erosion profiles relative to implants formulated using the pure polymer (Figure 7). With preformed systems fabricated using bulk eroding polymers, the onset of erosion has been explained as an effect of the percolation threshold. In percolation theory, the polymer is treated as a lattice of interconnected points [54, 59]. Over time, as the oligomers are lost to the bath-side solution, the interconnectivity of the lattice is lost. This results in a critically low number of nodes within the lattice, and elevated pore interconnectivity [54, 59]. Erosion begins when the interconnected network of pores leads to the surface of the implant, which results in the rapid release of the internal degradation products to the solution. We hypothesize that this process is also occurring with the ISFIs [54]. Interestingly, the onset of erosion corresponds to both the final solidification of the depots, and the sharp increases in the rate of drug release, which we speculate to be a result of the percolation phenomenon. Polymer degradation occurred more rapidly than anticipated, which we speculate may be a result of the implant size, an effect of the residual solvent, or even the elevated surface area due to the highly interconnected porous interior. The rate kinetics increased with Mw, which is in agreement with the findings of Wu et al., who hypothesized that the elevated degradation kinetics were a result of an increased number of reaction sites on the larger polymers [60].
The effects of polymer Mw on release were more complex than anticipated. Burst release from implants appeared to be controlled by the polymer with the higher burst. While, diffusion release was anticipated to be an average of the polymers used, the release profile instead appeared to closely follow the behavior of the polymer with the higher Mw. Finally degradation facilitated release appeared to be controlled by the lower Mw polymer. It was also observed that as the polymer ratio approached 10:1, intermediate behavior was lost (data not shown). After the degradation phase of release, a lockdown occurred in all implants, which we hypothesize may be a result of latent crystallization of the implants as a result of polymer degradation, resulting in a decrease in diffusivity and an increase in affinity for the drug and polymer [61].
Due to the high initial rate of phase inversion relative to drug release, a nonlinear relationship was developed to correlate drug release and phase inversion [50]. A rapid transition to linear behavior occurred for all polymer formulations except for implants containing the 15 kDa PLGA (Figure 10). When implants were formed in vivo, the nonlinearity was lost, and both phase inversion and release occurred rapidly. These changes may occur as a consequence of reactive forces generated by the injection of the implant into the tissue space displacing the tissue. This displacement results in the formation of reactive forces that then act on the implant leading to elevated loss of solvent and drug [26]. The elevated rate of release has previously been observed [26], but the use of the 15kDa:53kDa polymer blend resulted in a statistically lower overall release when compared with previous studies using 29kDa PLGA implants. These findings may be a consequence of the lower initial swelling observed with the polymer blend.
Characterization of ISFI has traditionally been performed using dark ground imaging systems capable of tracking both the diffusion fronts of water into the implant as well as the polymer precipitation [12-14, 46]. While this technique provides a wealth of information, it cannot be used in vivo. Techniques such as electron paramagnetic spectroscopy have been used to track changes in the microenvironment viscosity, providing phase inversion data both in vivo and in vitro, but this technique is not used to generate actual images of the implants [17, 18]. The use of medical imaging to characterize implants is compelling due to the wealth of information that can be obtained. The use of nuclear imaging techniques such as single-photon emission computed tomography (SPECT) or positron emission tomography (PET) may provide a means by which the drug distribution can be characterized in vivo with tremendous sensitivity, but may be somewhat limited due to the poor spatial resolution. Fluorescence imaging techniques such as fluorescence molecular tomography (FMT) provides an additional avenue by which the spatial distribution of drug may be obtained, with improved spatial resolution relative to PET/SPECT. MRI has previously been used to monitor solvent exchange [18], and provides an additional method by which implants can be evaluated in situ. While no single modality can be used to completely characterize these implants, the combination of these techniques can provide comprehensive information that may be used to elucidate the underlying parameters that control how implants will behave in a variety of environments.
Conclusion
ISFIs provide a unique platform which is simple to manufacture, has a diverse range of potential applications, and can be administered through a minimally invasive injection and monitored using medical imaging. In this study, molar ratios of the polymer used in the implant formulation were altered so that the effects of matrix composition on implant behavior could provide an alternative means by which the release from these depots can be tailored to fit a desired application. Results confirmed that intermediate implant behavior could be achieved by altering the molar ratio of the polymers used in the depots. Evaluation of polymer erosion data along with concurrent visualization of the implants provided insight into the longitudinal effect of autocatalysis and changes in the outer shell diffusivity on drug release, swelling, and phase inversion. Noninvasive characterization of implants using ultrasound imaging was able to provide unique insight into the behavior of these systems in situ, which may not only be used to evaluate ISFI systems, but may be extended to preformed polymer systems as well. Advances in these techniques such as the ability to monitor the implants in 4 dimensions (X,Y,Z, and time), may substantially improve characterization. Currently, the parameters that alter the release kinetics in vivo are poorly understood, but through the use of medical imaging, the multivariable system may be simplified. Evaluation of solvent exchange using MRI, or SPECT/PET analysis to evaluate macrophage activity may lead to improved understanding as to what effect the host has on implant behavior in situ.
Acknowledgements
This work was supported by NIH grant R01CA118399 to AAE and T32EB007509-01 to LS. The authors would like to thank Casey Johnson for his technical assistance in this project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Exner AA, Saidel GM. Drug-eluting polymer implants in cancer therapy. Expert Opinion on Drug Delivery. 2008;5:775-88
2. Langer R. Implantable Controlled Release Systems. Pharmacology & Therapeutics. 1983;21:35-51
3. Saltzman WM, Fung LK. Polymeric implants for cancer chemotherapy. Adv Drug Deliv Rev. 1997;26:209-30
4. Liu QF, Zhang H, Zhou GC, Xie SB, Zou H, Yu YA. et al. In vitro and in vivo study of thymosin alpha1 biodegradable in situ forming poly(lactide-co-glycolide) implants. International Journal of Pharmaceutics. 2010;397:122-9
5. Solorio L, Patel RB, Wu H, Krupka T, Exner AA. Advances in Image-Guided Intratumoral Drug Delivery Techniques. Therapeutic Delivery. 2010;1:307-22
6. Dunn RL, English JP, Cowsar DR, Vanderbilt DP. Biodegradable in situ forming implants and methods of producing the same. US. 1990
7. Dunn RL, Garrett S. The drug delivery and biomaterial attributes of the ATRIGEL technology in the treatment of periodontal disease. Expert opinion on investigational drugs. 1998;7:1483-91
8. Dunn RL, Tipton AJ. Polymeric compositions useful as controlled release implants. US. 1997
9. Gad HA, El-Nabarawi MA, El-Hady SSA. Formulation and Evaluation of PLA and PLGA In Situ Implants Containing Secnidazole and/or Doxycycline for Treatment of Periodontitis. Aaps Pharmscitech. 2008;9:878-84
10. Ravivarapu HB, Moyer KL, Dunn RL. Sustained suppression of pituitary-gonadal axis with an injectable, in situ forming implant of leuprolide acetate. J Pharm Sci. 2000;89:732-41 doi:10.1002/(SICI)1520-6017(200006)89:6<732::AID-JPS4>3.0.CO;2-D [pii] 10.1002/(SICI)1520-6017(200006)89:6<732::AID-JPS4>3.0.CO;2-D
11. Ravivarapu HB, Moyer KL, Dunn RL. Parameters affecting the efficacy of a sustained release polymeric implant of leuprolide. Int J Pharm. 2000;194:181-91 doi:S0378517399003713 [pii]
12. Brodbeck KJ, DesNoyer JR, McHugh AJ. Phase inversion dynamics of PLGA solutions related to drug delivery. Part II. The role of solution thermodynamics and bath-side mass transfer. J Control Release. 1999;62:333-44 doi:S0168365999001595 [pii]
13. Graham PD, Brodbeck KJ, McHugh AJ. Phase inversion dynamics of PLGA solutions related to drug delivery. J Control Release. 1999;58:233-45 doi:S0168365998001588 [pii]
14. Mchugh AJ, Tsay CS. Dynamics of the Phase Inversion Process. Journal of Applied Polymer Science. 1992;46:2011-21
15. Ravivarapu HB, Moyer KL, Dunn RL. Sustained activity and release of leuprolide acetate from an in situ forming polymeric implant. AAPS PharmSciTech. 2000;1:E1. doi:10.1208/pt010101
16. Smolders CA, Reuvers AJ, Boom RM, Wienk IM. Microstructures in Phase-Inversion Membranes.1. Formation of Macrovoids. Journal of Membrane Science. 1992;73:259-75
17. Kempe S, Metz H, Mader K. Do in situ forming PLG/NMP implants behave similar in vitro and in vivo? A non-invasive and quantitative EPR investigation on the mechanisms of the implant formation process. J Control Release. 2008;130:220-5
18. Kempe S, Metz H, Pereira PGC, Mader K. Non-invasive in vivo evaluation of in situ forming PLGA implants by benchtop magnetic resonance imaging (BT-MRI) and EPR spectroscopy. European Journal of Pharmaceutics and Biopharmaceutics. 2010;74:102-8
19. McHugh AJ, Graham PD, Brodbeck KJ. Phase inversion dynamics of PLGA solutions related to drug delivery. Biomedical Materials-Drug Delivery, Implants and Tissue Engineering. 1999;550:41-6376
20. Packhaeuser CB, Schnieders J, Oster CG, Kissel T. In situ forming parenteral drug delivery systems: an overview. Eur J Pharm Biopharm. 2004;58:445-55 doi:10.1016/j.ejpb.2004.03.003S0939641104000463 [pii]
21. Patel RB, Carlson AN, Solorio L, Exner AA. Characterization of formulation parameters affecting low molecular weight drug release from in situ forming drug delivery systems. Journal of Biomedical Materials Research Part A. 2010;94A:476-84
22. Solorio L, Babin BM, Patel RB, Mach J, Azar N, Exner AA. Noninvasive characterization of in situ forming implants using diagnostic ultrasound. Journal of Controlled Release. 2010;143:183-90
23. Jeon O, Krebs M, Alsberg E. Controlled and sustained gene delivery from injectable, porous PLGA scaffolds. J Biomed Mater Res A. 2011;98:72-9
24. Krebs MD, Sutter KA, Lin AS, Guldberg RE, Alsberg E. Injectable poly(lactic-co-glycolic) acid scaffolds with in situ pore formation for tissue engineering. Acta Biomater. 2009;5:2847-59
25. Krupka TM, Weinberg BD, Ziats NP, Haaga JR, Exner AA. Injectable polymer depot combined with radiofrequency ablation for treatment of experimental carcinoma in rat. Invest Radiol. 2006;41:890-7
26. Patel RB, Solorio L, Wu HP, Krupka T, Exner AA. Effect of injection site on in situ implant formation and drug release in vivo. Journal of Controlled Release. 2010;147:350-8
27. Singh S, Singh J. Phase-sensitive polymer-based controlled delivery systems of leuprolide acetate: In vitro release, biocompatibility, and in vivo absorption in rabbits. International Journal of Pharmaceutics. 2007;328:42-8
28. Al-Tahami K, Oak M, Singh J. Controlled delivery of basal insulin from phase-sensitive polymeric systems after subcutaneous administration: In vitro release, stability, biocompatibility, in vivo absorption, and bioactivity of insulin. J Pharm Sci.
29. Astaneh R, Erfan M, Barzin J, Mobedi H, Moghimi H. Effects of Ethyl Benzoate on Performance, Morphology, and Erosion of PLGA Implants Formed In Situ. Advances in Polymer Technology. 2008;27:17-26
30. Astaneh R, Erfan M, Moghimi H, Mobedi H. Changes in morphology of in situ forming PLGA implant prepared by different polymer molecular weight and its effect on release behavior. J Pharm Sci. 2009;98:135-45
31. Bakhshi R, Vasheghani-Farahani E, Mobedi H, Jamshidi A, Khakpour M. The effect of additives on naltrexone hydrochloride release and solvent removal rate from an injectable in situ forming PLGA implant. Polymers for Advanced Technologies. 2006;17:354-9
32. Brodbeck KJ, Pushpala S, McHugh AJ. Sustained release of human growth hormone from PLGA solution depots. Pharm Res. 1999;16:1825-9
33. Chhabra S, Sachdeva V, Singh S. Influence of end groups on in vitro release and biological activity of lysozyme from a phase-sensitive smart polymer-based in situ gel forming controlled release drug delivery system. International Journal of Pharmaceutics. 2007;342:72-7
34. DesNoyer JR, McHugh AJ. Role of crystallization in the phase inversion dynamics and protein release kinetics of injectable drug delivery systems. J Control Release. 2001;70:285-94 doi:S0168365900003540 [pii]
35. DesNoyer JR, McHugh AJ. The effect of Pluronic on the protein release kinetics of an injectable drug delivery system. J Control Release. 2003;86:15-24 doi:S0168365902002936 [pii]
36. Kranz H, Yilmaz E, Brazeau GA, Bodmeier R. In vitro and in vivo drug release from a novel in situ forming drug delivery system. Pharmaceutical Research. 2008;25:1347-54
37. Liu H, Venkatraman SS. Effect of Polymer Type on the Dynamics of Phase Inversion and Drug Release in Injectable In Situ Gelling Systems. J Biomater Sci Polym Ed. 2012
38. Liu Y, Kemmer A, Keim K, Curdy C, Petersen H, Kissel T. Poly(ethylene carbonate) as a surface-eroding biomaterial for in situ forming parenteral drug delivery systems: A feasibility study. European Journal of Pharmaceutics and Biopharmaceutics. 2010;76:222-9
39. Luan X, Bodmeier R. In situ forming microparticle system for controlled delivery of leuprolide acetate: influence of the formulation and processing parameters. Eur J Pharm Sci. 2006;27:143-9
40. Luan X, Bodmeier R. Influence of the poly(lactide-co-glycolide) type on the leuprolide release from in situ forming microparticle systems. J Control Release. 2006;110:266-72
41. Yapar A, Baykara T. Effects of Solvent Combinations on Drug Release from Injectable Phase Sensitive Liquid Implants. Turk J Pharm Sci. 2010;7:49-56
42. Zare M, Mobedi H, Barzin J, Mivehchi H, Jamshidi A, Mashayekhi R. Effect of additives on release profile of leuprolide acetate in an in situ forming controlled-release system: In vitro study. Journal of Applied Polymer Science. 2008;107:3781-7
43. Singh S, Singh J. Controlled release of a model protein lysozyme from phase sensitive smart polymer systems. International Journal of Pharmaceutics. 2004;271:189-96
44. Wang LW, Kleiner L, Venkatraman S. Structure formation in injectable poly(lactide-co-glycolide) depots. Journal of Controlled Release. 2003;90:345-54
45. Hatefi A, Amsden B. Biodegradable injectable in situ forming drug delivery systems. J Control Release. 2002;80:9-28 doi:S0168365902000081 [pii]
46. Mchugh AJ, Miller DC. The Dynamics of Diffusion and Gel Growth during Nonsolvent-Induced Phase Inversion of Polyethersulfone. Journal of Membrane Science. 1995;105:121-36
47. Astaneh R, Erfan M, Mobedi H, Moghimi HR. Formulation of an Injectable Implant for Peptide Delivery and Studying the Effect of Polymer Molecular Weight on Its Release Behavior. Journal of Peptide Science. 2004;10:142
48. Eliaz RE, Wallach D, Kost J. Delivery of soluble tumor necrosis factor receptor from in-situ forming PLGA implants: in-vivo. Pharm Res. 2000;17:1546-50
49. McHugh AJ. The role of polymer membrane formation in sustained release drug delivery systems. J Control Release. 2005;109:211-21 doi:S0168-3659(05)00493-1 [pii]10.1016/j.jconrel.2005.09.038
50. Solorio L, Olear AM, Zhou H, Beiswenger AC, Exner AA. Effect of cargo properties on in situ forming implant behavior determined by noninvasive ultrasound imaging. Drug Deliv Transl Res. 2012;2:45-55
51. Barton BF, Reeve JL, McHugh AJ. Observations on the dynamics of nonsolvent-induced phase inversion. Journal of Polymer Science Part B-Polymer Physics. 1997;35:569-85
52. Li DF, Chung TS, Ren JZ, Wang R. Thickness dependence of macrovoid evolution in wet phase-inversion asymmetric membranes. Industrial & Engineering Chemistry Research. 2004;43:1553-6
53. Vogrin N, Stropnik C, Musil V, Brumen M. The wet phase separation: the effect of cast solution thickness on the appearance of macrovoids in the membrane forming ternary cellulose acetate/acetone/water system. Journal of Membrane Science. 2002;207:139-41
54. Gopferich A. Polymer bulk erosion. Macromolecules. 1997;30:2598-604
55. Li S, Vert M. Hydrolytic degradation of the coral/poly(DL-lactic acid) bioresorbable material. J Biomater Sci Polym Ed. 1996;7:817-27
56. von Burkersroda F, Schedl L, Gopferich A. Why degradable polymers undergo surface erosion or bulk erosion. Biomaterials. 2002;23:4221-31
57. Alexis F. Factors affecting the degradation and drug-release mechanism of poly(lactic acid) and poly[(lactic acid)-co-(glycolic acid)]. Polymer International. 2005;54:36-46
58. Solorio L, Solorio LD, Gleeson S, Olear AM, Carlson AC, Exner AA. Phase Inverting Polymer Systems In Drug Delivery And Medicine. Ehlers TP, Wilhelm JK, editors. Polymer Phase Behavior: Nova Science Publishers, Inc. 2011
59. Leuenberger H, Rohera BD, Haas C. Percolation Theory - a Novel-Approach to Solid Dosage Form Design. International Journal of Pharmaceutics. 1987;38:109-15
60. Wu XS, Wang N. Synthesis, characterization, biodegradation, and drug delivery application of biodegradable lactic/glycolic acid polymers. Part II: Biodegradation. Journal of Biomaterials Science-Polymer Edition. 2001;12:21-34
61. Hurrell S, Cameron RE. The effect of initial polymer morphology on the degradation and drug release from polyglycolide. Biomaterials. 2002;23:2401-9
Author contact
![]() Corresponding author: Exner A.A., Department of Radiology, Case Center for Imaging Research, Case Western Reserve University, 11100 Euclid Ave., Cleveland, OH 44106. Phone: 216-844-3544 fax: 216-844-5922 e-mail: agata.exneredu.
Corresponding author: Exner A.A., Department of Radiology, Case Center for Imaging Research, Case Western Reserve University, 11100 Euclid Ave., Cleveland, OH 44106. Phone: 216-844-3544 fax: 216-844-5922 e-mail: agata.exneredu.
Citation styles
APA
Solorio, L., Olear, A.M., Hamilton, J.I., Patel, R.B., Beiswenger, A.C., Wallace, J.E., Zhou, H., Exner, A.A. (2012). Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging. Theranostics, 2(11), 1064-1077. https://doi.org/10.7150/thno.4181.
ACS
Solorio, L.; Olear, A.M.; Hamilton, J.I.; Patel, R.B.; Beiswenger, A.C.; Wallace, J.E.; Zhou, H.; Exner, A.A. Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging. Theranostics 2012, 2 (11), 1064-1077. DOI: 10.7150/thno.4181.
NLM
Solorio L, Olear AM, Hamilton JI, Patel RB, Beiswenger AC, Wallace JE, Zhou H, Exner AA. Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging. Theranostics 2012; 2(11):1064-1077. doi:10.7150/thno.4181. https://www.thno.org/v02p1064.htm
CSE
Solorio L, Olear AM, Hamilton JI, Patel RB, Beiswenger AC, Wallace JE, Zhou H, Exner AA. 2012. Noninvasive Characterization of the Effect of Varying PLGA Molecular Weight Blends on In Situ Forming Implant Behavior Using Ultrasound Imaging. Theranostics. 2(11):1064-1077.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.