Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2012; 2(8):757-768. doi:10.7150/thno.4756 This issue Cite
Research Paper
Gold Nanorods Conjugated with Doxorubicin and cRGD for Combined Anticancer Drug Delivery and PET Imaging
Yuling Xiao1, Hao Hong2,3, Vyara Z. Matson4, Alireza Javadi1, Wenjin Xu1, Yunan Yang2, Yin Zhang3, Jonathan W. Engle3, Robert J. Nickles2,3, Weibo Cai2,3 ![]() , Douglas A. Steeber4, Shaoqin Gong1
, Douglas A. Steeber4, Shaoqin Gong1 ![]()
1. Department of Biomedical Engineering and Wisconsin Institutes for Discovery, University of Wisconsin-Madison, Madison, WI 53706, USA;
2. Department of Radiology, University of Wisconsin-Madison, Madison, WI 53705, USA;
3. Department of Medical Physics, University of Wisconsin-Madison, Madison, WI 53705, USA;
4. Department of Biological Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA.
Received 2012-6-18; Accepted 2012-7-11; Published 2012-8-6
Abstract
A multifunctional gold nanorod (GNR)-based nanoplatform for targeted anticancer drug delivery and positron emission tomography (PET) imaging of tumors was developed and characterized. An anti-cancer drug (i.e., doxorubicin (DOX)) was covalently conjugated onto PEGylated (PEG: polyethylene glycol) GNR nanocarriers via a hydrazone bond to achieve pH-sensitive controlled drug release. Tumor-targeting ligands (i.e., the cyclo(Arg-Gly-Asp-D-Phe-Cys) peptides, cRGD) and 64Cu-chelators (i.e., 1,4,7-triazacyclononane-N, N', N''-triacetic acid (NOTA)) were conjugated onto the distal ends of the PEG arms to achieve active tumor-targeting and PET imaging, respectively. Based on flow cytometry analysis, cRGD-conjugated nanocarriers (i.e., GNR-DOX-cRGD) exhibited a higher cellular uptake and cytotoxicity than non-targeted ones (i.e., GNR-DOX) in vitro. However, GNR-DOX-cRGD and GNR-DOX nanocarriers had similar in vivo biodistribution according to in vivo PET imaging and biodistribution studies. Due to the unique optical properties of GNRs, this multifunctional GNR-based nanoplatform can potentially be optimized for combined cancer therapies (chemotherapy and photothermal therapy) and multimodality imaging (PET, optical, X-ray computed tomography (CT), etc.).
Keywords: Gold nanorod (GNR), drug delivery, nanoparticles, cancer, positron emission tomography (PET), cRGD peptide, tumor targeting
Introduction
Cancer continues to be one of the world's most devastating diseases, with more than 10 million new cases each year [1-3]. Current cancer treatments include surgical intervention, radiation therapy, and chemotherapy which face formidable challenges as they often cause collateral, debilitating, and adverse effects to the patients [4]. Nanotechnology provides extraordinary opportunities to address these challenges as nanocarriers can offer both passive and active targeting abilities towards cancerous tissues/cells [5-7]. Passive tumor targeting ability is attributed to the enhanced permeability and retention (EPR) effect exhibited by tumor tissues which allows the nanocarriers to preferentially accumulate in the tumor tissue [3, 5]. Active tumor-targeting ability can be achieved by conjugating certain tumor-targeting ligands such as peptides, antibodies, and aptamers that can recognize and bind specifically to receptors that are overexpressed by cancer cells [5, 7-9]. Moreover, many of the pharmacologic properties of free drugs including poor water solubility and in vivo stability can be improved by nanocarriers. Drug nanocarriers can also provide a stimuli-responsive drug release profile at the pathological site thereby optimizing the pharmacokinetics of the drug and enhancing the therapeutic efficacy [3, 5-7, 10-16].
Over the past two decades, great strides have been made in the design and application of nanomedicine. A number of therapeutic nanocarriers have already been approved for clinical use [3, 17]. The current focus of nanomedicine is to develop multifunctional tumor-targeting drug/agent nanocarriers, including those capable of co-delivering anti-cancer drugs and imaging contrast agents which are termed cancer theranostics (i.e., combined therapy and diagnostics), thereby providing a more effective and multimodal approach to cancer management [6, 7, 18-20].
Molecular imaging has been extensively studied for cancer detection and staging, as well as the evaluation of the in vivo biodistribution of various nanoparticles/nanocarriers [21-23]. Among them, positron emission tomography (PET) imaging has become increasingly popular in both preclinical and clinical settings as it offers excellent sensitivity for deep tissues, higher detection efficiency, and non-invasiveness [24, 25]. Nevertheless, there is not a single modality that is sufficient to collect all necessary information for a particular assessment. For example, PET imaging has poor spatial resolution. Thus, multimodality imaging is often applied in clinical oncology [22, 24-27]. Recently, optical imaging has emerged as a complement to radionuclide-based imaging techniques as it offers convenient, non-invasive, non-radioactive, real-time, and high-resolution imaging of diseased tissues [21, 28]. Particularly, near-infrared (NIR; 700-1,000 nm) fluorescence imaging is potentially a very useful secondary imaging modality to complement PET imaging as NIR light can penetrate several centimeters of tissue. This is because light absorption by endogenous chromophores in native tissue is minimized in the NIR region [21, 28, 29]. Gold nanorods (GNRs) with suitable aspect ratios can absorb and scatter strongly in the NIR region, which can be used for enhanced optical imaging and photothermal cancer therapy [30-33]. In fact, due to its tunable optical properties (dependent upon the aspect ratio) and chemical versatility, GNRs have been explored for a broad spectrum of biomedical applications including drug/gene delivery and photothermal therapy, as well as computed tomography (CT) and/or optical imaging [21, 30-32, 34-36].
Herein, we report a multifunctional and water-soluble GNR-based nanoplatform that integrates chemotherapy, PET imaging, and molecular targeting into one system (Figure 1). Doxorubicin (DOX), a model anticancer drug, was conjugated onto the GNR via a pH-sensitive hydrazone bond to achieve a pH-responsive drug release profile. A cyclic arginine-glycine-aspartic acid (cRGD) peptide was conjugated selectively to the poly(ethylene glycol) (PEG) terminal ends, which can specifically bind to integrin αvβ3 that is overexpressed on sprouting tumor vessels and many tumor cells [37-39]. 1,4,7-triazacyclononane-N, N', N''-triacetic acid (NOTA), a macrocyclic 64Cu (t1/2: 12.7 h) chelator, was selectively conjugated onto the distal ends of the PEG arms for PET imaging, which is used to non-invasively and quantitatively monitor the in vivo biodistribution and tumor-targeting efficacy of the GNR-based nanocarriers.
A schematic illustration of the multifunctional GNR-DOX-cRGD nanocarriers for tumor-targeted drug delivery and PET imaging.
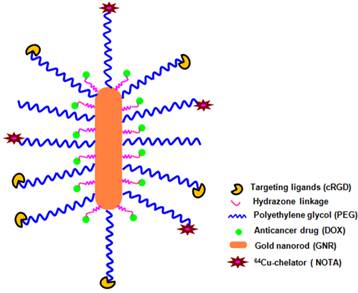
Materials and methods
Materials
HAuCl4.3H2O (99.9%), NaBH4 (99%), L-ascorbic acid (AA, 99+%), cetyltrimethylammonium bromide (CTAB, 99%), AgNO3 (99+%), anhydrous dimethyl sulfoxide (DMSO), aminoethanethiol hydrochloride (AET.HCl), methyl thioglycolate (MTG), anhydrous hydrazine, 4-dimethylamino pyridine (DMAP), N-hydroxysuccinimide (NHS), and 1,3-dicyclohexylcarbodiimide (DCC) were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and used without further purification. Triethylamine (TEA) and dimethylformamide (DMF) were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and were distilled before use. The heterobifunctional PEG derivative, R (R = maleimide or methoxy)-PEG114-NHS (Mw: 5000) was purchased from JenKem Technology (Allen, TX, USA). The anti-cancer drug, doxorubicin·HCl (DOX), was purchased from Tecoland Corporation (Irvine, CA, USA). Cyclo(Arg-Gly-Asp-D-Phe-Cys) (c(RGDfC)) peptide was purchased from Peptides International (Louisville, KY, USA). p-SCN-Bn-NOTA was purchased from Macrocyclics, Inc. (Dallas, TX, USA). Dulbecco's Modified Eagle Medium (DMEM) was purchased from Gibco BRL (Carlsbad, CA, USA). U87MG human glioblastoma cells (expressing high levels of integrin αvβ3 [40]) were purchased from ATCC and were cultured in DMEM supplemented with 10% fetal calf serum. All other chemicals and reagents used were of analytical reagent grade. Phosphate and acetate buffered solutions were prepared in our laboratory. Ultrapure deionized water (DI water, Milli-Q Water Systems) was used for all solution preparations and experiments. Glassware used for preparing GNRs was cleaned by soaking in aqua regia followed by washing with DI water. During 64Cu-labeling, water and all buffers used were of Millipore grade and pre-treated with Chelex 100 resin (50-100 mesh, Sigma-Aldrich, St. Louis, MO, USA) to ensure that the aqueous solution was heavy-metal free. PD-10 desalting columns were purchased from GE Healthcare (Piscataway, NJ, USA). 64Cu was produced via a 64Ni(p,n)64Cu reaction using a cyclotron at the University of Wisconsin-Madison [41].
Synthesis of bare GNRs
Bare GNRs were prepared via the well-established two-step seed-mediated growth method with a slight modification [42, 43]. Briefly, 600 µL of ice-cold 10 mM NaBH4 solution was quickly added to 250 µL of 0.01 M auric acid (HAuCl4) dissolved in a 0.1 M CTAB surfactant solution. The solution was stirred for 2 min. Afterwards, it was allowed to react for 2 h to form the CTAB-capped Au nanoparticles to be used as seeds for the synthesis of GNRs. To synthesize the GNRs, 95 mL of CTAB solution (0.1 M) was added to a 200 mL flask. Afterwards, 950 µL of AgNO3 solution (0.01 M) mixed in 5 mL of HAuCl4 solution (0.01 M) was added to the CTAB solution. Subsequently, 550 µL of freshly prepared AA solution (0.1 M) were added to the above mixture as a weak reducing agent. After swirling for 3 min, the gold color disappeared which indicated the reduction of Gold (III) to Gold (I). Lastly, 100 µL of CTAB-capped Au nanoparticle seeds were added. The solution was left standing overnight. The resulting GNRs were separated from the reaction solution via centrifugation at 14,000 rpm for 12 minutes, washed with deionized water twice to remove any residual reactants, and separated from water via centrifugation at 14,000 rpm for about 15 minutes using a Beckman Coulter Avanti J-26-XPI centrifuge (Beckman, Germany).
Synthesis of methyl thioglycolate (MTG) and aminoethanethiol (AET) conjugated GNRs
In order to conjugate PEG and DOX onto the GNR, MTG and AET linkers were first anchored to the surface of the GNR. The feed molar ratio of GNR: MTG: AET was 1: 2,000: 2,000. 20 µL of the MTG and AET mixture (both at 1 mM) was added to 10 mL of 1 nM GNRs in a mixture of water and methanol solution (in 1:1 molar ratio) and the resulting mixture was allowed to react for 12 h in order to form stable thiol monolayers. Thereafter, the reaction mixture was centrifuged at 14,000 rpm for 15 min and re-dispersed in 10 mL of DI water.
PEGylation of the MTG- and AET-conjugated GNRs
A mixture of NHS esters, methoxy-PEG-NHS (denoted as “MPEG-NHS”; Mw: 5,000) and maleimide-PEG-NHS (denoted as “Mal-PEG-NHS”; Mw: 5,000), was reacted with the MTG- and AET-conjugated GNRs. The molar ratio of GNRs: (MPEG-NHS + Mal-PEG-NHS) was set at 1: 2,000 with the molar ratio of MPEG-NHS: Mal-PEG-NHS set at 2:3. The reaction was conducted in DI water at room temperature for 6 h after 30 min of N2 bubbling. After the reaction was complete, the solution was purified by dialysis against DI water for 2 days. The solution was then filtered through a 0.22 µm membrane followed by freeze-drying.
Conjugation of DOX onto the functionalized GNRs
DOX was conjugated onto the GNRs via a two-step reaction. During the first step, the methoxy groups of MTG were substituted with anhydrous hydrazide by an ester-amide exchange aminolysis reaction in anhydrous DMSO at 40 oC for 24 h, with the molar ratio of GNR: hydrazide set at 1: 2,000. The resulting hydrazine-modified GNRs were purified by extensive dialysis against 0.25% ammonia solution for 48 h followed by freeze-drying. In the second step, DOX was conjugated to the hydrazide group of the GNRs through a pH-sensitive hydrazone linkage. The reaction was conducted in DMSO solution at room temperature for 24 h with a 1.5-fold excess amount of DOX to hydrazide in the dark. The unbound DOX was removed completely by dialysis against DI water in the dark.
Conjugation of cRGD and NOTA onto the functionalized GNRs
cRGD and NOTA conjugation onto the functionalized GNRs were achieved through the reaction between the Mal groups on the surface of GNRs with the thiol groups of cRGD and/or thiol-functionalized NOTA (NOTA-SH). NOTA-SH was prepared by reacting p-SCN-Bn-NOTA with AET in the presence of TEA. The reaction was allowed to stay in DI water at room temperature for 5 h under N2 atmosphere. Thereafter, a predetermined amount of cRGD and NOTA-SH aqueous solution was added into the solution of PEGylated, DOX-conjugated GNRs suspended in water with the feed molar ratio of cRGD: NOTA: PEGylated DOX-conjugated GNRs at 400: 200: 1. The molar ratio for cRGD: NOTA was set at 2:1, with the total molar amount equaling that of Mal-PEG. This reaction was carried out in PBS buffer (pH 7.4) at room temperature for 5 h under N2 atmosphere. After the reaction was complete, the mixture was purified by dialysis against DI water for 48 h.
Determination of the DOX loading content
The DOX loading content (DLC), defined as the weight percentage of DOX in the GNR nanocarriers, was quantified by UV-Vis analysis. First, DOX was released completely from the nanocarriers in a solution of 0.1 N HCl to cleave the hydrazone bonds. The absorbance of DOX at 485 nm was measured to determine the DLC in the solution using a previously established calibration curve. The DLC measurements were performed in triplicate for each sample.
Characterization of the morphology of the GNRs and GNR nanocarriers
The UV-Vis absorption spectra of the GNR seed and GNR solutions were recorded on a Labtech 1100 UV-Vis spectrophotometer. Zeta potential of the GNRs was determined by dynamic light scattering (DLS) (ZetaSizer Nano ZS90, Malvern Instrument, USA). Samples for transmission electron microscopy (TEM, LEO912-OMEGA, Zeiss, Germany) analysis were prepared by drying a dispersion of the GNRs on a copper grid coated with amorphous carbon.
Evaluation of the pH-controlled drug release profiles
The pH-responsive DOX release behavior of the GNR nanocarriers was studied in three mediums with different pH values by UV-spectrophotometry. Briefly, 10 mg of freeze-dried cRGD/DOX-conjugated GNRs were dispersed in 5 mL of medium (10 mM phosphate buffer [pH 7.4] or acetate buffer [pH 5.3 or pH 6.6]) and placed in a dialysis bag with a Mw cut-off of 2 kDa. Subsequently, the dialysis bag was immersed in 95 mL of the same medium and kept in a horizontal laboratory shaker at 37 oC under constant shaking. At selected time intervals, 5 mL of the buffered solution outside of the dialysis bag was removed for UV analysis and replaced by fresh buffer solution of the same volume. The amount of released DOX was quantified by UV measurement at 485 nm. Each sample was measured in triplicate.
Cellular uptake study
Cellular uptake of the GNR nanocarriers was analyzed using flow cytometry. U87MG cells (1 × 105) were seeded in 24-well culture plates and cultured in DMEM overnight. The cells were then treated with free DOX, GNR-DOX-cRGD (i.e., targeted), or GNR-DOX (i.e., non-targeted) for 15 and 120 min (DOX concentration: 10 µg/mL). Untreated cells served as a negative control for background fluorescence. Thereafter, the cells were lifted using CellstripperTM (Media Tech, Inc., Manassas, VA) and washed. DOX uptake was analyzed using a FACSCalibur flow cytometer and CellQuest™ Pro software (BD Bioscience, San Jose, CA). An average of 1 × 104 cells was analyzed from each sample with the DOX fluorescence intensity shown on a four-decade log scale. Cellular uptake experiments were repeated three times with differences in the mean DOX fluorescence between treatment groups analyzed for significance (p < 0.05) using a two-tailed Student's t-test.
Cytotoxicity evaluation
Cytotoxicity of the GNR nanocarriers to U87MG cells was studied using the MTT assay. First, U87MG cells (5 × 104) were incubated in suspension for 2 h in DMEM containing free DOX, GNR-DOX-cRGD, or GNR-DOX (all with a 10 μg/mL of DOX concentration) or control media. Thereafter, the cells were washed and plated in triplicate in 96-well plates and cultured for 48 h. The cells were then incubated for 4 h with DMEM containing 250 µg/mL of MTT. After discarding the culture medium, 200 µL of DMSO was added to dissolve the precipitates, and the resulting solution was measured for absorbance at 570 nm with a reference wavelength of 690 nm using a microtiter plate reader (Molecular Devices, Sunnyvale, CA). Significant differences in cytotoxicity between treatment groups were determined using a two-tailed Student's t-test with p < 0.05 being considered significant.
Animal model
All animal studies were conducted under a protocol approved by the University of Wisconsin Institutional Animal Care and Use Committee. U87MG cells were used for tumor inoculation when they reached ~80% confluence. Four- to five-week-old female athymic nude mice were purchased from Harlan (Indianapolis, IN, USA) and tumors were established by subcutaneously injecting 5 × 106 cells, suspended in 100 μL of 1:1 mixture of medium and Matrigel (BD Biosciences, Franklin lakes, NJ, USA), into the front flank of the mice [44]. The tumor sizes were monitored every other day and the animals were subjected to in vivo experiments when the diameter of the tumors reached 6~8 mm (typically 4-5 weeks after inoculation).
64Cu-labeling of the GNR nanocarriers
Both targeted (i.e., GNR-DOX-cRGD) and non-targeted (i.e., GNR-DOX) nanocarriers have NOTA selectively conjugated onto the distal ends of the PEG arms allowing for 64Cu-labeling. 64CuCl2 (74~185 MBq) was diluted in 300 μL of 0.1 M sodium acetate buffer (pH 6.5) and added to a solution of GNR-DOX-cRGD or GNR-DOX (pre-centrifuged at 5,000 rpm for 5 min to remove possible aggregates) with the ratio of ~0.2 μg of NOTA per 37 MBq of 64CuCl2. The reaction mixture was incubated for 30 min at 42 ºC with constant shaking. GNR-DOX-64Cu and GNR-DOX-cRGD-64Cu were purified using PD-10 columns with PBS as the mobile phase. The radioactive fractions containing GNR-DOX-64Cu or GNR-DOX-cRGD-64Cu were collected for in vivo experiments.
MicroPET imaging
PET scans were performed using a microPET/microCT Inveon rodent model scanner (Siemens Medical Solutions USA, Inc.). Each U87MG tumor-bearing mouse was injected with 5-10 MBq of the tracer via tail vein, and 5-15 min static PET scans were performed at various time points post-injection (p.i.). The images were reconstructed using a maximum a posteriori (MAP) algorithm with no attenuation or scatter correction [41]. For each microPET scan, three-dimensional (3D) regions-of-interest (ROIs) were drawn over the tumor and major organs by using vendor software (Inveon Research Workshop; IRW) on decay-corrected whole-body images. Assuming a tissue density of 1 g/mL, the ROIs were converted to MBq/g using a conversion factor (pre-determined using a 50 mL centrifuge tube filled with ~50 MBq 64CuCl2 as a phantom) and then divided by the administered radioactivity to obtain quantitative data in the unit of percentage of injected dose per gram of tissue (%ID/g). After the PET scans, the mice were euthanized for biodistribution studies.
Biodistribution studies
Ex vivo biodistribution studies were carried out to confirm that the quantitative tracer uptake values based on non-invasive microPET imaging truly represented the actual tracer distribution in tumor-bearing mice. After the last PET scans at 48 h p.i., the mice were euthanized and blood, U87MG tumors, and major organs/tissues were collected and wet-weighed. The radioactivity in the tissue was measured using a gamma-counter (Perkin Elmer) and presented as %ID/g (mean ± SD). A second group of mice were injected with GNR-DOX-64Cu or GNR-DOX-cRGD-64Cu and euthanized at 5 h p.i. (when the tumor uptake was prominent) for biodistribution studies.
Results and Discussion
Synthesis and characterization of the GNR nanocarriers
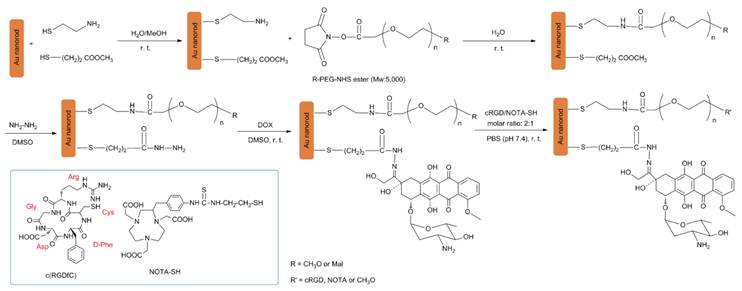
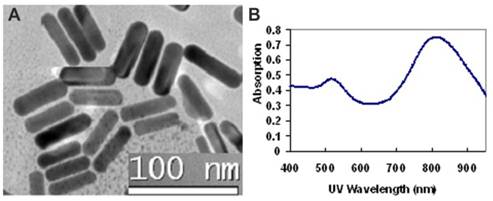
The GNR nanocarriers were prepared according to Scheme 1 (Figure S1). First, bare (i.e., un-functionalized) GNRs were synthesized according to the well-established seed-mediated growth method [42, 43]. It was found that keeping a constant temperature at 28 oC throughout the GNR synthesis was vital for the formation of uniform GNRs. The UV/IR absorption spectrum and a TEM image of the bare GNRs are shown in Figure 2. The bare GNRs had a length and diameter of approximately 45 and 10 nm, respectively (Figure 2A), and exhibited two absorption bands: a weak short-wavelength band around 520 nm (due to the transverse electronic oscillation) and a strong long-wavelength band around 810 nm (due to the longitudinal oscillation of electrons) (Figure 2B). It is well-known that NIR light can be transmitted deep into the tissue without causing any significant damage. As such, GNRs can be potentially used for photothermal therapy. The zeta potential was determined to be 42 mV due to the existence of the cationic surfactant (i.e., CTAB) which was used for capping and stabilizing the GNRs during synthesis.
(Scheme 1) The synthetic scheme for the multifunctional GNR-DOX-cRGD-64Cu nanocarriers.
The morphology and optical properties of bare GNRs: (A) A TEM micrograph showing the GNRs with an aspect ratio of 4.5; (B) Optical absorption spectrum showing two surface plasmon resonance peaks at 520 nm and 810 nm.
In order to functionalize the GNRs, a mixture of two thiolated linkers (i.e., MTG and AET) was anchored onto the GNRs through the thiol moiety. MTG linkers were employed for DOX conjugation while the AET linkers were used to conjugate the heterofunctional NHS ester of Mal-PEG-NHS, which is used for cRGD and NOTA conjugation. MPEG-NHS was also conjugated to the AET linkers in order to adjust the solubility and stability of the resulting GNR nanocarriers in physiological conditions, as well as to help achieve the desired cRGD and NOTA molar ratios. Specifically, the molar ratio of MTG and AET was set at 1:1 in order to maximize the DOX loading level while retaining the desired solubility and stability of the GNRs.
The average number of MTG and AET arms attached to a given GNR was estimated as follows: the volume of one GNR, assuming 45 nm for the length and 10 nm for the diameter (based on TEM images), is V = πr2l = π × (5 nm)2 × 45 nm = 3,534.3 nm3, which means that the theoretical average mass per GNR is 6.82 × 10-17 g (ρAu=19,300 kg m-3). Hence, the Mw for 1 mole of GNRs is M = m × NA ≅ 41,076,000 g mol-1 = 4.11 × 107 g mol-1 (NA = Avogadro constant). The increase in mass after the coupling of MTG and AET is about 0.45%, which was measured by comparing the weight of the freeze-dried GNRs before and after MTG and AET conjugation in triplicate, indicating that the molecular weight of the MTG and AET functionalized GNR rose to 41,260,007 g mol-1. As such, the average MTG and AET mass per mole of GNRs is about 184,007 g. According to the average molecular weight of the AET and MTG linkers ((77+106)/2=91.5 g mol-1) and the MTG and AET mass per mole of GNRs, the number of arms per GNR was estimated to be 184,007/91.5 = 2011 MTG and AET arms per GNR. Therefore, the number of MTG and AET anchored onto each GNR was ~1,000 each.
A mixture of Mal-PEG-NHS and MPEG-NHS was coupled onto the AET linkers at the molar ratio of MPEG-NHS:Mal-PEG-NHS = 2:3 via a reaction between activated NHS esters and primary amines. For each individual GNR, a maximum of 600 arms of Mal-PEG were used to conjugate cRGD and NOTA while a maximum of 400 arms of MPEG were used to adjust the solubility/stability in physiological conditions of the GNR nanocarriers. Subsequently, DOX was conjugated through an acid-sensitive hydrazone linkage via a two-step reaction [10]. Lastly, cRGD and NOTA were conjugated onto the distal ends of the MAL-PEG arms through the thiol groups. DOX conjugation on non-targeted GNR nanocarriers (i.e., GNR-DOX) was also prepared in a similar fashion to be used as a control.
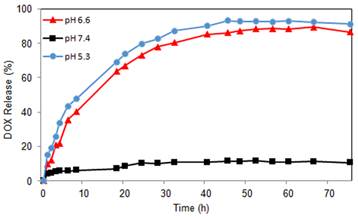
The amount of DOX incorporated into the GNR nanocarriers was determined by UV analysis after cleaving the hydrazone linkers between the nanocarriers and DOX. The DLC was calculated to be 0.84 wt.%. In order to test the pH-sensitivity of the GNR nanocarriers, in vitro drug release studies were performed under simulated physiological conditions at pH 5.3, 6.6, and 7.4 at 37 oC. As shown in Figure 3, the pH value of the medium had a strong effect on the DOX release rate from the GNR nanocarriers. The drug release at pH 7.4 was negligible with an initial burst release of about 6% and a total of 11% drug release after 45 h. However, at pH values of 5.3 and 6.6, the DOX release rate was much faster, with approximately 93% and 86% of the drug released within 45 h, respectively, showing that DOX release from the GNR nanocarriers in an acidic environment was governed by the acid-cleavable hydrazone linkage. As such, the pH-sensitive GNR nanocarriers will minimize premature drug release during circulation in the bloodstream (pH 7.4), yet provide a sufficient amount of drug to effectively kill the cancer cells once the GNR nanocarriers are internalized into the endocytic compartments where the pH value ranges from 4.5 to 6.5. This will greatly enhance the tumor-directed therapeutic efficacy while minimizing non-specific systemic spread of toxicity. These findings agree with our previous studies on intracellular environment-sensitive drug delivery systems [45, 46].
DOX release profiles of GNR-DOX-cRGD at pH 5.3, 6.6, and 7.4.
Cellular uptake and distribution of the GNR nanocarriers
It is well-known that the inclusion of targeting ligands providing specific nanoparticle-cell surface interactions is crucial in determining the ultimate location of the nanoparticle. Integrin αvβ3, an important biomarker overexpressed in sprouting tumor vessels and many tumor cells, plays a key role in endothelial cell survival during angiogenesis [27, 47-49]. Hereby, cRGD peptide, a potent integrin αvβ3 antagonist, was chosen as a targeting molecule for this study. To investigate the cellular uptake of the GNR nanocarriers, flow cytometry was performed using the U87MG human glioblastoma cell line which takes advantage of the intrinsic fluorescence of DOX. Figure 4A shows representative quantitative flow cytometry results of the cellular uptake of the GNR-DOX-cRGD and GNR-DOX nanocarriers compared to free DOX and untreated control cells at early (15 min) and late (120 min) time points. As expected, the negative control cells without any DOX treatment showed only a low level of autofluorescence at both time points. After 15 min, U87MG cells treated with free DOX showed the highest level of fluorescence, which was 2-fold higher (p < 0.005) than cells treated with GNR-DOX-cRGD (Figure 4B). At this time point, GNR-DOX nanocarriers showed the same level of fluorescence as the control cells, indicating negligible uptake of non-targeted GNRs. Overall, these results showed that conjugating cRGD to GNRs increased their cellular uptake by 2.2-fold, even at a very early time point.
Flow cytometry analysis of U87MG cells treated with free DOX, GNR-DOX-cRGD, GNR-DOX (DOX concentration: 10 μg/mL), or medium alone (control) for 15 and 120 min at 37 ºC. (A) Representative histogram plots showing DOX fluorescence following cellular uptake. (B) Summary data of flow cytometry results. Bars represent the mean fluorescence intensity (MFI) values ± SEM of DOX from three independent experiments. * indicates that values for the 120 min time point were significantly different from the 15 min time point; p < 0.02.
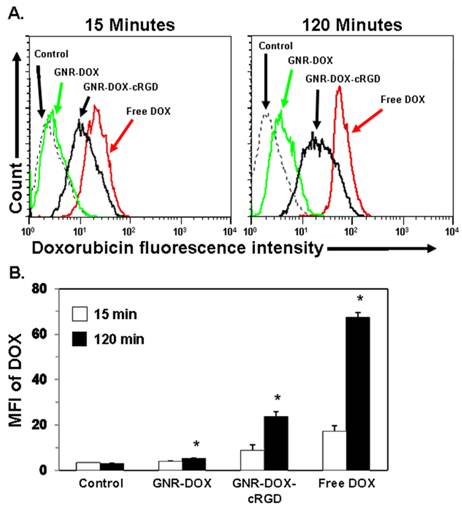
After 120 min of incubation, cells treated with free DOX still exhibited the highest level of fluorescence, increasing by 3.9-fold over the 15 min time point (p < 0.005). Similarly, cells treated with the GNR-DOX-cRGD showed a significant 2.7-fold increase in fluorescence over the earlier time point (p < 0.005). In contrast, cells treated with the GNR-DOX showed only a minimal, albeit significant (p < 0.02), increase in fluorescence (by 1.3-fold) over the 15 min time point. Importantly, at this late time point the GNR-DOX-cRGD showed a 4.5-fold increase in uptake compared to the GNR-DOX, which was even greater than that observed at the 15 min time point (p < 0.02). Thus, targeting αVβ3 integrin expressed on U87MG cells is clearly an effective means of increasing the uptake of the GNR nanocarriers into these cells. Since cRGD-conjugated GNRs are taken up by receptor-mediated endocytosis, it is not unexpected that their uptake was lower than that of free DOX, which readily diffuses across the cell membrane.
Cytotoxicity of the GNR nanocarriers
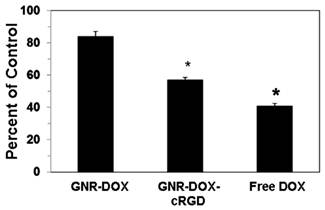
To determine the effectiveness of the GNR nanocarriers in mediating cytotoxicity against U87MG tumor cells, cells were treated for 2 h with GNR-DOX-cRGD, GNR-DOX, or free DOX (all at 10 μg/mL DOX concentration), cultured for 48 h, and analyzed using the MTT assay. Figure 5 shows that treatment of U87MG cells with GNR-DOX did not significantly reduce the viability of the cells relative to control cells. In contrast, GNR-DOX-cRGD significantly decreased the viability down to 57% of the control cells. Free DOX demonstrated the highest level of cytotoxicity (41% of control). These results are consistent with the flow cytometry data described above and demonstrate that the level of cellular uptake of the GNR nanocarriers or free DOX directly correlated with cytotoxicity. Therefore, increasing cell uptake using cRGD peptides conjugated onto the GNR nanocarriers to target αVβ3 integrin on tumor cells can directly increase cellular destruction.
Cytotoxicity of U87MG cells treated for 2 h with free DOX, GNR-DOX-cRGD, or GNR-DOX (all at 10 μg/mL DOX concentration). Following the 2 h treatment, the cells were cultured for 48 h and the degree of cytotoxicity was determined using the MTT assay. * indicates that values for the cytotoxicity of DOX and GNR-DOX-cRGD were significantly different from the control; p < 0.01.
MicroPET studies
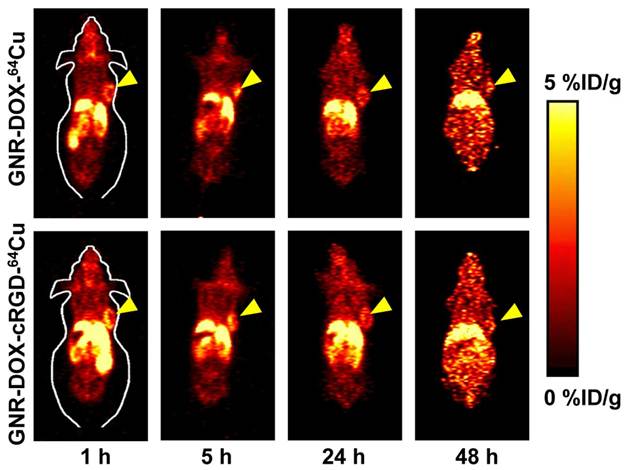
64Cu-labeling, including final purification using a PD-10 column, took 80 ± 10 min (n = 10). The decay-corrected radiochemical yield was about 20-30%. For serial PET scans, the time points of 1, 5, 24, and 48 h p.i. were chosen based on our previous experience with other 64Cu-labeled nanomaterials [50, 51]. The coronal slices that contain the U87MG tumors are shown in Figure 6. The quantitative data obtained from ROI analysis are shown in Figure 7A.
In vivo investigation of 64Cu-labeled GNR nanocarriers. Serial coronal PET images of U87MG tumor-bearing mice at various time points post-injection of GNR-DOX-cRGD-64Cu or GNR-DOX-64Cu. Images are representative of 3 mice per group and arrowheads indicate the U87MG tumors.
ROI analysis and biodistribution studies. (A) Time-activity curves of the U87MG tumor, liver, blood, and muscle upon intravenous injection of GNR-DOX-cRGD-64Cu or GNR-DOX-64Cu (n = 3). (B) Biodistribution of GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu in U87MG tumor-bearing mice at 5 h post-injection (n = 3). (C) Biodistribution of GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu in U87MG tumor-bearing mice at 48 h post-injection (n = 3).
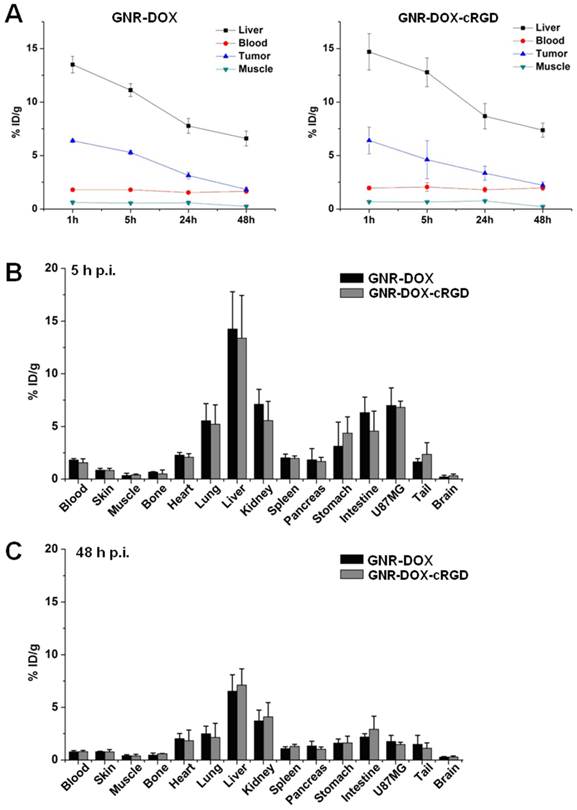
Similar to other radiolabeled nanoparticles, the uptake of GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu in the liver (due to the uptake in the reticuloendothelial system (RES), hepatic clearance, and possible trans-chelation of 64Cu) was prominent at early time points and gradually declined over time. The radioactivity in the blood pool was relatively low (~2 %ID/g after 1 h p.i.), indicating that the circulation half-lives of the GNRs were quite short. The liver uptake of GNR-DOX-RGD-64Cu was 14.7 ± 1.7, 12.7 ± 1.3, 8.7 ± 1.2, and 7.4 ± 0.6 %ID/g at 1, 5, 24, and 48 h p.i., respectively, while the radioactivity in the blood was 1.9 ± 0.2, 2.0 ± 0.4, 1.8 ± 0.2, and 2.0 ± 0.2 %ID/g at 1, 5, 24, and 48 h p.i., respectively (n = 3, Figure 7A). The tumor uptake of GNR-DOX-RGD-64Cu was clearly visible even at 1 h p.i. and gradually decreased afterwards (6.4 ± 1.2, 4.6 ± 1.8, 3.3 ± 0.7, and 2.2 ± 0.3 %ID/g at 1, 5, 24, and 48 h p.i., respectively; n = 3; Figure 7A).
For GNR-DOX-64Cu, the liver uptake was 13.5 ± 0.8, 11.1 ± 0.6, 7.8 ± 0.7, and 6.6 ± 0.7 %ID/g at 1, 5, 24, and 48 h p.i., respectively, and the radioactivity in the blood was 1.8 ± 0.2, 1.8 ± 0.1, 1.5 ± 0.1, and 1.7 ± 0.2 %ID/g at 1, 5, 24, and 48 h p.i., respectively (n = 3, Figure 7A). The tumor uptake of GNR-DOX-64Cu was 6.4 ± 0.2, 5.3 ± 0.2, 3.1 ± 0.3, and 1.8 ± 0.2 %ID/g at 1, 5, 24, and 48 h p.i., respectively (n = 3; Figure 7A). These data suggested that conjugating cRGD onto the GNR nanocarriers did not confer a significant advantage in tumor uptake in vivo over the passive targeting of GNR nanocarriers due to the EPR effect.
Biodistribution studies
After the last PET scans at 48 h p.i., the mice were euthanized. The tissues were collected for biodistribution to further validate the in vivo PET data (Figure 7C). Separate groups of mice were injected with GNR-DOX-cRGD-64Cu or GNR-DOX-64Cu and euthanized at 5 h p.i. for biodistribution studies, when the tumor uptake of the radiolabeled GNR nanocarriers was prominent (Figure 7B). At 5 h p.i., the liver had the highest radioactivity uptake followed by the tumors, kidneys, and intestines. Comparing the biodistribution data at 5 h and 48 h p.i., uptake in all tissues dropped significantly, including in the tumors, which suggested that the uptake of both GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu nanocarriers was largely based on the EPR effect rather than the cRGD-integrin αvβ3 interaction, as tracer uptake based on specific targeting tends to exhibit durable tumor retention [50, 51]. Nonetheless, good tumor contrast was observed at all time points examined. The tumor/muscle ratio of GNR-DOX-cRGD-64Cu was 16.6 ± 1.2 and 3.6 ± 0.4 at 5 h and 48 h p.i., respectively (n = 3).
Similar to what was observed in the PET studies, comparison of the biodistribution data of GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu at 48 h p.i. revealed a corroborating distribution pattern for GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu nanocarriers in vivo. Overall, the quantification results obtained from biodistribution studies and PET scans matched very well, confirming that quantitative ROI analysis of non-invasive microPET scans truly reflected the distribution of PET tracers in vivo.
The challenges of achieving efficient in vivo tumor targeting using nanocarriers conjugated with active tumor-targeting ligands such as antibodies and peptides have been well-documented in the literature [7, 52]. In this study, although in vitro experiments clearly demonstrated an increase in integrin αvβ3-mediated uptake of GNR-DOX-cRGD over GNR-DOX in U87MG cells, there was no significant difference in the U87MG tumor uptake in vivo between GNR-DOX-cRGD-64Cu and GNR-DOX-64Cu nanocarriers. In cell-based studies, the U87MG cells are directly incubated with GNR-DOX-cRGD and there is virtually no barrier for RGD-integrin αvβ3 binding. However, the in vivo biodistribution of the functionalized GNRs at the tumor site is affected by many factors in vivo because of their complex interactions with the biological system. A recent study reported that while certain types of targeting peptides indeed increased the tumor accumulation of the GNRs, cRGD actually reduced the tumor accumulation of the GNRs 2-3 fold, which was attributed to a shorter circulation time and a higher uptake in the liver and spleen for the cRGD-conjugated GNRs [53]. The type of targeting ligands also affected the nanoparticle distribution in tumor cells and tumor microenvironment [53]. In addition, our previous studies on cRGD-conjugated superparamagnetic iron oxide nanocarriers and cRGD-conjugated unimolecular micelles had clearly shown that cRGD-conjugated nanoparticles had a much higher tumor accumulation than their corresponding non-targeted nanoparticles [45, 46]. As such, both the type of the nanoparticles and the type of targeting peptides can affect the in vivo tumor accumulation of these nanoparticles.
Conclusions
Multifunctional water-soluble GNR nanocarriers were developed to provide both tumor-targeted drug delivery and PET imaging. These GNR nanocarriers exhibit pH-sensitive drug release behaviour, which can minimize non-specific systemic spread of toxic drugs during circulation while maximizing the efficiency of tumor-targeted anticancer drug delivery. Cellular uptake and cytotoxicity studies demonstrated that the cRGD-conjugated GNR nanocarriers showed a higher cellular uptake and thus a more potent cytotoxicity in cancer cells overexpressing αVβ3 integrin when compared to the cRGD-free GNR nanocarriers. Although the cRGD-conjugated GNR nanocarriers did not show any significant increases in tumor accumulation according to both the in vivo PET imaging and biodistribution studies, this study does provide a novel nanoplatform for possible integration of multi-functionality including molecular targeting, chemotherapy, photothermal therapy, as well as multimodality imaging such as PET/optical, which can potentially lead to improved therapeutic efficacy and cancer monitoring. Future work may focus on a further understanding of the optimal conditions for combined photothermal therapy and chemotherapy, as well as dual-modality PET and optical imaging utilizing these multifunctional GNR nanocarriers.
Acknowledgements
This work is supported, in part, by the National Science Foundation (CMMI 1032186), the University of Wisconsin Carbone Cancer Center, the University of Wisconsin-Madison and the University of Wisconsin-Milwaukee Intercampus grant, the Department of Defense (W81XWH-11-1-0644 and W81XWH-11-1-0648), and the NIH through the UW Radiological Sciences Training Program 5 T32 CA009206-32. We also would like to thank Phillip B. Gonzales in the Materials Science Program at the University of Wisconsin-Madison for editing the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
2. Jemal A, Siegel R, Xu J, Ward E. Cancer Statistics, 2010. CA Cancer J Clin. 2010;60:277-300
3. Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nano. 2007;2:751-60
4. Chabner BA, Roberts TG Jr. Timeline: Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65-72
5. Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153:198-205
6. Cabral H, Nishiyama N, Kataoka K. Supramolecular nanodevices: from design validation to theranostic nanomedicine. Acc Chem Res. 2011;44:999-1008
7. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161-71
8. Yu MK, Park J, Jon S. Targeting strategies for multifunctional nanoparticles in cancer imaging and therapy. Theranostics. 2012;2:3-44
9. Huang P, Xu C, Lin J, Wang C, Wang X, Zhang C. et al. Folic acid-conjugated graphene oxide loaded with photosensitizers for targeting photodynamic therapy. Theranostics. 2011;1:240-50
10. Bae Y, Jang WD, Nishiyama N, Fukushima S, Kataoka K. Multifunctional polymeric micelles with folate-mediated cancer cell targeting and pH-triggered drug releasing properties for active intracellular drug delivery. Mol Biosyst. 2005;1:242-50
11. Bae Y, Kataoka K. Significant enhancement of antitumor activity and bioavailability of intracellular pH-sensitive polymeric micelles by folate conjugation. J Control Release. 2006;116:e49-50
12. Farrell D, Alper J, Ptak K, Panaro NJ, Grodzinski P, Barker AD. Recent advances from the national cancer institute alliance for nanotechnology in cancer. ACS Nano. 2010;4:589-94
13. Gunasekera UA, Pankhurst QA, Douek M. Imaging applications of nanotechnology in cancer. Target Oncol. 2009;4:169-81
14. Guo S, Qiao Y, Wang W, He H, Deng L, Xing J. et al. Poly([varepsilon]-caprolactone)-graft-poly(2-(N, N-dimethylamino) ethyl methacrylate) nanoparticles: pH dependent thermo-sensitive multifunctional carriers for gene and drug delivery. J Mater Chem. 2010;20:6935-41
15. Yong KT, Roy I, Swihart MT, Prasad PN. Multifunctional nanoparticles as biocompatible targeted probes for human cancer diagnosis and therapy. J Mater Chem. 2009;19:4655-72
16. Patra CR, Bhattacharya R, Mukherjee P. Fabrication and functional characterization of goldnanoconjugates for potential application in ovarian cancer. J Mater Chem. 2010;20:547-54
17. Petros RA, DeSimone JM. Strategies in the design of nanoparticles for therapeutic applications. Nat Rev Drug Discov. 2010;9:615-27
18. Kim K, Kim JH, Park H, Kim YS, Park K, Nam H. et al. Tumor-homing multifunctional nanoparticles for cancer theragnosis: simultaneous diagnosis, drug delivery, and therapeutic monitoring. J Control Release. 2010;146:219-27
19. Rosi NL, Mirkin CA. Nanostructures in biodiagnostics. Chem Rev. 2005;105:1547-62
20. Bhirde AA, Liu G, Jin A, Iglesias-Bartolome R, Sousa AA, Leapman RD. et al. Combining portable raman probes with nanotubes for theranostic applications. Theranostics. 2011;1:310-21
21. Choi J, Yang J, Park J, Kim E, Suh J-S, Huh Y-M. et al. Specific near-IR absorption imaging of glioblastomas using integrin-targeting gold nanorods. Adv Funct Mater. 2011;21:1082-8
22. Zhang F, Zhu L, Liu G, Hida N, Lu G, Eden HS. et al. Multimodality imaging of tumor response to doxil. Theranostics. 2011;1:302-9
23. Kessinger CW, Togao O, Khemtong C, Huang G, Takahashi M, Gao JM. Investigation of in vivo targeting kinetics of αvβ3-specific superparamagnetic nanoprobes by time-resolved MRI. Theranostics. 2011;1:263-73
24. Hong H, Zhang Y, Sun J, Cai W. Molecular imaging and therapy of cancer with radiolabeled nanoparticles. Nano Today. 2009;4:399-413
25. Townsend DW. Dual-Modality Imaging: Combining Anatomy and Function. J Nucl Med. 2008;49:938-55
26. Jennings LE, Long NJ. 'Two is better than one'-probes for dual-modality molecular imaging. Chem Commun. 2009;28:3511-24
27. Zhang Y, Yang Y, Cai W. Multimodality imaging of integrin αvβ3 expression. Theranostics. 2011;1:135-48
28. Joshi BP, Wang TD. Exogenous molecular probes for targeted imaging in cancer: focus on multi-modal imaging. Cancers (Basel). 2010;2:1251-87
29. Weissleder R. A clearer vision for in vivo imaging. Nat Biotechnol. 2001;19:316-7
30. Huang X, El-Sayed IH, Qian W, El-Sayed MA. Cancer cell imaging and photothermal therapy in the near-infrared region by using gold nanorods. J Am Chem Soc. 2006;128:2115-20
31. Kuo W-S, Chang C-N, Chang Y-T, Yang M-H, Chien Y-H, Chen S-J. et al. Gold nanorods in photodynamic therapy, as hyperthermia agents, and in near-infrared optical imaging. Angew Chem Int Ed. 2010;49:2711-5
32. Murphy CJ, Gole AM, Hunyadi SE, Stone JW, Sisco PN, Alkilany A. et al. Chemical sensing and imaging with metallic nanorods. Chem Commun (Camb). 2008:544-57
33. Ntziachristos V, Bremer C, Weissleder R. Fluorescence imaging with near-infrared light: new technological advances that enable in vivo molecular imaging. Eur Radiol. 2003;13:195-208
34. Chakravarthy KV, Bonoiu AC, Davis WG, Ranjan P, Ding H, Hu R. et al. Gold nanorod delivery of an ssRNA immune activator inhibits pandemic H1N1 influenza viral replication. Proc Natl Acad Sci U S A. 2010;107:10172-7
35. Huang H-C, Rege K, Heys JJ. Spatiotemporal Temperature Distribution and cancer cell death in response to extracellular hyperthermia induced by gold nanorods. ACS Nano. 2010;4:2892-900
36. von Maltzahn G, Centrone A, Park J-H, Ramanathan R, Sailor MJ, Hatton TA. et al. SERS-coded gold nanorods as a multifunctional platform for densely multiplexed near-infrared imaging and photothermal heating. Adv Mater. 2009;21:3175-80
37. Nasongkla N, Shuai X, Ai H, Weinberg BD, Pink J, Boothman DA. et al. cRGD-functionalized polymer micelles for targeted doxorubicin delivery. Angew Chem Int Ed Engl. 2004;43:6323-7
38. Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491-7
39. Chen X. Integrin targeted imaging and therapy. Theranostics. 2011;2011:28-9
40. Cai W, Shin DW, Chen K, Gheysens O, Cao Q, Wang SX. et al. Peptide-labeled near-infrared quantum dots for imaging tumor vasculature in living subjects. Nano Lett. 2006;6:669-76
41. Hong H, Yang Y, Zhang Y, Engle JW, Barnhart TE, Nickles RJ. et al. Positron emission tomography imaging of CD105 expression during tumor angiogenesis. Eur J Nucl Med Mol Imaging. 2011;38:1335-43
42. Nikoobakht B, El-Sayed MA. Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem Mater. 2003;15:1957-62
43. Sau TK, Murphy CJ. Seeded high yield synthesis of short Au nanorods in aqueous solution. Langmuir. 2004;20:6414-20
44. Cai W, Wu Y, Chen K, Cao Q, Tice DA, Chen X. In vitro and In vivo Characterization of 64Cu-Labeled AbegrinTM, a humanized monoclonal antibody against integrin αvβ3. Cancer Res. 2006;66:9673-81
45. Xiao Y, Hong H, Javadi A, Engle JW, Xu W, Yang Y. et al. Multifunctional unimolecular micelles for cancer-targeted drug delivery and positron emission tomography imaging. Biomaterials. 2012;33:3071-82
46. Yang X, Hong H, Grailer JJ, Rowland IJ, Javadi A, Hurley SA. et al. cRGD-functionalized, DOX-conjugated, and 64Cu-labeled superparamagnetic iron oxide nanoparticles for targeted anticancer drug delivery and PET/MR imaging. Biomaterials. 2011;32:4151-60
47. Brooks PC, Montgomery AMP, Rosenfeld M, Reisfeld RA, Hu T, Klier G. et al. Integrin αvβ3 antagonists promote tumor regression by inducing apoptosis of angiogenic blood vessels. Cell. 1994;79:1157-64
48. Niu G, Chen X. Why integrin as a primary target for imaging and therapy. Theranostics. 2011;1:30-47
49. Chen K, Chen X. Integrin targeted delivery of chemotherapeutics. Theranostics. 2011;1:189-200
50. Cai W, Chen K, Li ZB, Gambhir SS, Chen X. Dual-function probe for PET and near-infrared fluorescence imaging of tumor vasculature. J Nucl Med. 2007;48:1862-70
51. Liu Z, Cai W, He L, Nakayama N, Chen K, Sun X. et al. In vivo biodistribution and highly efficient tumour targeting of carbon nanotubes in mice. Nat Nanotechnol. 2007;2:47-52
52. Cai W, Chen X. Nanoplatforms for targeted molecular imaging in living subjects. Small. 2007;3:1840-54
53. Huang X, Peng X, Wang Y, Wang Y, Shin DM, El-Sayed MA. et al. A reexamination of active and passive tumor targeting by using rod-shaped gold nanocrystals and covalently conjugated peptide ligands. ACS Nano. 2010;4:5887-96
Author contact
![]() Corresponding author: Weibo Cai, PhD, Department of Radiology, University of Wisconsin-Madison, Madison, WI 53705, USA. Tel: 608-262-1749; Email: WCaiorg. Shaoqin Gong, PhD, Department of Biomedical Engineering and Wisconsin Institutes for Discovery, University of Wisconsin-Madison, Madison, WI 53706, USA. Tel: 608-316-4311; Email: sgongwisc.edu.
Corresponding author: Weibo Cai, PhD, Department of Radiology, University of Wisconsin-Madison, Madison, WI 53705, USA. Tel: 608-262-1749; Email: WCaiorg. Shaoqin Gong, PhD, Department of Biomedical Engineering and Wisconsin Institutes for Discovery, University of Wisconsin-Madison, Madison, WI 53706, USA. Tel: 608-316-4311; Email: sgongwisc.edu.