Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results and Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2012; 2(6):618-628. doi:10.7150/thno.4561 This issue Cite
Research Paper
DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells
Jing Ruan, Hua Song, Chao Li, Chenchen Bao, Hualin Fu, Kan Wang, Jian Ni, Daxiang Cui ![]()
Department of Bio-Nano Science and Engineering, Key Laboratory for Thin Film and Microfabrication of Ministry of Education, Research Institute of Micro/Nano Science and Technology, Shanghai Jiao Tong University, Shanghai200240, P. R. China.
Received 2012-5-6; Accepted 2012-6-1; Published 2012-6-15
Citation:
Ruan J, Song H, Li C, Bao C, Fu H, Wang K, Ni J, Cui D. DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells. Theranostics 2012; 2(6):618-628. doi:10.7150/thno.4561. https://www.thno.org/v02p0618.htm
Other stylesAbstract
Embryonic stem (ES) cells have great potential in applications such as disease modeling, pharmacological screening and stem cell therapies. Up to date, there is no related report on the use of ES cells as tracking and contrast reagents of cancer cells in vivo. Herein we report that DiR-labeled murine ES cells can recognize and target gastric cancer cells in vivo. DiR-labeled murine ES (mES) cells (5×106) were intravenously injected into gastric tumor-bearing mice. The biodistribution of DiR-labeled mES cells was monitored by IVIS imaging within 24 h. Major organs were harvested and analyzed by immunofluorescence staining and Western blotting. Chemotaxis assay was employed to investigate the chemotaxis of ES cells tracking cancer cells. Fluorescent imaging results showed that DiR-labeled mES cells targeted gastric cancer tissue in vivo as early as 10 min post-injection, reaching a peak at 2h post-injection. Immunofluorescence staining and Western blotting results showed gastric cancer tissues specifically expressed SSEA-1. In vitro migration tests confirmed that mES cells actively moved to test sites with different concentration of CXCL12 in a dose-dependent manner. In conclusion, DiR-labeled mES cells may be used for gastric cancer targeted imaging in vivo, and have great potential in applications such as identifying and imaging of early gastric cancer in near future.
Keywords: murine embryonic stem cells, gastric cancer cells, target imaging, migration, chemotaxis.
Introduction
Since the firstly isolation of the murine embryonic stem (mES) cells in the 1980s, ES cells show their pluripotent nature, differentiating into cell types of all primary germ lineages [1-3]. In recent years, researchers have paid more and more attention on the in vivo monitoring and therapy potential of stem cells [4-6]. Generally, stem cell therapies consist of two aspects, one is the in vivo targeted migration of stem cells to the target tissues, and another is the proliferation and differentiation of stem cells in the target tissues to regenerate new organs [7-10]. Those applications have promising prospects in regenerative medicine.
Stomach malignancy is currently the fourteenth most common cancer in the United States and the second most common cancer in China [11, 12]. Gastric cancer is still the second most common cause of cancer-related death in the world. It remains difficult to cure effectively even in Western countries, primarily because most patients are identified at the advanced stages of the malignancy [13]. Thus, early recognition and tracking of gastric cancer cells in vivo would be of particular significance. Our group has tried to establish an early gastric cancer pre-warning system since 2005 [14]. We hoped to find early gastric cancer cells in vivo by multi-mode targeted imaging techniques [15-18]. However, our efforts were stalled by a lack of specific gastric cancer biomarkers. This is one reason why the search for an alternative way to recognize and track early gastric cancer cells in vivo has become a central subject in this field.
Among all the imaging techniques, near-infrared (NIR) imaging and bioluminescence imaging (BLI) have become the most popular modalities [19, 20]. NIR imaging has many advantages over other imaging means, because it can penetrate biological tissues such as skin and blood more efficiently than visible light [21, 22]. BLI combined with NIR light has been found to enable real-time observation of stem cell trafficking and in vivo gene transfer.
DiR dye is a lipophilic, NIR fluorescent cyanine dye ideal for staining cytoplasmic membrane. The two long 18-carbon chains of DiR dye can insert into the cell membrane, resulting in specific and stable cell staining with negligible dye transfer between cells. The NIR property of DiR dye makes it ideal for in vivo imaging because of significantly reduced autofluorescence from the animal at higher wavelength [23]. In this study, mES cells were labeled with NIR DiR dyes, in vivo distribution of DiR-labeled mES cells (DiR-mES cells) was monitored by an IVIS imaging system, and the chemotaxis mechanism of ES cells tracking cancer cells was investigated. The significant finding is that tumor tissues or metastatic cancer cells in vivo can be tracked by using mES cells as tracking and contrast reagents.
Materials and Methods
All animal experiments (NO.SYXK2007-0025) were approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University.
Feeder-free cultured murine embryonic stem (mES) cells
Murine embryonic stem (GFP-SV129, mES) cells were provided by the Shanghai Institute of Digestive Disease, Renji Hospital. The mES cells were cultured with completed medium, which is composed of Knockout-DMEM (Gibco) supplemented with fetal bovine serum (FBS, Gibco), non-essential amino acids (NEAA, Gibco), L-glutamine (Gibco), β-mercaptoethanol (Gibco) and recombinant human leukemia inhibitory factor (LIF, Chemicon). The mES cells were routinely passaged every 2 days, and the medium was changed on alternate days. The feeder-free mES cells were prepared by using the direct transition method: mES cells were split onto newly gelatinized plates without feeders and incubated for 30 min, and then the supernatant culture medium were collected and transferred into newly gelatinized plates without feeders and cultured until mitosis. The mES cells were continuously cultured for three to four or more splits by this method to eliminate all feeders.
Labeling mES cells with DiR and in vitro cell imaging
The mES cells were washed three times with PBS, trypsinized with 0.05% trypsin-ethylene diaminetetra acetic acid (EDTA; Gibco-Invitrogen). The mES cells were incubated with 3.5 μg/mL DiR buffer for 30 min at 37°C according to the protocol of XenoLight DiR (Caliper Lifesciences). DiR-labeled mES cells were used as the DiR(+) test group, mES cells which were not incubated with DiR were washed with PBS (pH 7.0) and marked with DiR(-) control group. Then the DiR(+) and DiR(-) cells were centrifuged for 3 min at 1000 rpm and 4°C, and washed twice with PBS buffer and examined for viability using a Typan Blue Staining Cell Viability Assay Kit (Beyotime). Finally, DiR(+) and DiR(-) cells(5×106) were resuspended in 0.2 mL PBS buffer, and performed fluorescence imaging in a 96-well black culture dish by IVIS system under 710 nm of excitation and 760 nm of emission. The DiR(+) and DiR(-) cells were continuously cultured and imaged at 4, 8, 12, and 24 h after first imaging under the same imaging conditions. The intensity of the region of interest (ROI) was plotted in units of the maximum number of photons per second per centimeter square per steridian (p/s/cm2/sr). In order to evaluate the changes of the fluorescent signals varied cell numbers, 1×106, 1×105, 1×104, 1×103 and 1×102 DiR(+) cells were also counted and performed fluorescent imaging in a 96-well black culture dish by IVIS system under 710 nm of excitation and 760 nm of emission, the correlation between the number of DiR(+) cells and the intensity of fluorescent signals was plotted.
Preparation of bioluminescence gastric cancer mouse models
Male athymic nude mice (4 weeks old, 17 ± 2 g) were obtained from the Shanghai LAC Laboratory Animal Co. Ltd., Chinese Academy of Sciences (Shanghai, China, SCXK2007-0005). Bioluminescence mouse gastric cancer cell lines (MFC) (1x106) were injected subcutaneously into the right anterior flank areas of the mice. Four weeks later, tumors grew to approximately 5 mm in diameter, and bioluminescence images of gastric cancer mouse models were taken using an IVIS system with the exposure time of 30 s.
DiR-mES cells for targeted imaging of gastric cancer cells in vivo
DiR-mES cells (5×106) were intravenously injected into the gastric cancer mouse models (n=3) as test group. In control group, gastric cancer mouse models (n=3) were injected with DiR. These injected gastric cancer mouse models were monitored using the IVIS system at 0.5, 1, 2, 4, 6, 12, and 24h post-injection to obtain serial fluorescence images. The ideal filter conditions for DiR imaging were set at 710 nm of excitation and 760 nm of emission. Identical illumination settings (e.g., lamp voltage, filter, exposure time) were used in all animal imaging experiments. The mice were imaged at all-time points at lateral, supine and prone positions. Spleen, liver, lung, kidney, heart, and tumor tissues were collected for ex vivo imaging at 24 h post-injection. Gray-scale photographic images and fluorescent images of each sample were analyzed and overlaid using Living Image software (Xenogen, Alameda, CA). Regions of interest (ROI) were drawn over the signals, and average radiant efficiency was quantified in p/s/cm2/sr.
Immunofluorescence analysis and Western Blot analysis
For histological evaluation, the mice in the test and control groups were euthanized after imaging. Tumors and major organs (spleen, liver, lung, kidney, and heart) were excised, frozen, embedded, and sectioned into 8 µm slices. The expression of SSEA-1 in these tissues were examined under a fluorescence microscope (Olympus IX71) after performing immunohistochemical staining of slices. GFP imaging of ultra-thin slices were also imaged simultaneously. Meanwhile, the expression of SSEA-1 in tumor tissues were evaluated in protein level. Tumor tissues and important organs (liver, lung, kidney, spleen, and heart) were excised and lysed in protein lysis buffer. Equal amounts of sample lysates were separated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). The membranes were blocked with 0.1% BSA in Tris-buffered saline Tween-20 (TBST) buffer and incubated overnight at 4°C with anti-SSEA-1 antibody. The membranes were washed with TBST buffer and incubated with horseradish peroxidase-conjugated secondary antibodies. Enhanced chemiluminescence kits were used (Amersham, ECL kits).
Chemotaxis analysis
The number of CXCR4-positive cells among mES cells was analyzed using a flow cytometer. PE-labeled anti-CXCR4 antibody (BioLegend) was used to sort for CXCR4-positive cells. Amount of CXCL12 in the culture supernatants of MFC cells was examined by enzyme-linked immunosorbent assay (ELISA) with commercial kits (R & D System). The migration ability of mES cells was evaluated using a 48-well modified Boyden chamber. The polycarbonate filter (12 μm pore size, CN110416; Neuroprobe, Bethesda, MD) was precoated with fibronectin (5 μg/mL; Sigma Chemical, St Louis, MO). The mES cells were resuspended at 5×105/mL in the appropriate medium supplemented with 10% FBS and seeded in the upper chamber. Recombinant CXCL12 (R&D System) was used as a chemoattractant in the lower compartment. The chambers were incubated overnight at 37°C. Results were expressed as the percentage of migrating cells. Each experiment was performed in triplicate.
Statistical Analysis
All data were presented in this paper as means result ± S.D. Statistical differences were evaluated using the t-test and considered significance at P < 0.05 level.
Results and Discussion
Morphological observation of cultured mES cells
As shown in Fig.1A and 1C, GFP-SV 129 mES cells cultured on the feeder cells formed typical large colonies with long-ellipse or long-shuttle shape, mES cells exhibited small enchylema and big nucleoli with obvious stereoscopic traits and clear boundaries when cultured with feeder cells. It also had strong green fluorescence signals since this mES cell line carries the GFP marker, as shown in Fig.1B. Although mES cells in feeder-free layer also maintained a normal karyotype, remarkable changes in mES cell shape grown in a monolayer can be observed as shown in Fig.1D and 1F, the feeder-free mES cells also have strong green fluorescence signal (Fig.1E).
In vitro fluorescence imaging of DiR-mES cells
The NIR property of DiR dye makes it ideal for in vivo imaging. After the mES cells were labeled with DiR, DiR-mES cells (5×106) were resuspended and added into 96-well plate and performed fluorescence imaging at 0, 4, 8, 12, and 24 h, respectively. The results showed that the fluorescence signals of DiR(+) cells were stronger than that of DiR(-) cells. Although the fluorescent signals decreased over the time as shown in Fig.2A, it have statistical significance compared to the DiR(-) cells. Meanwhile, fluorescence imaging of different concentrations of DiR(+) cells were performed as shown in Fig.2B. The linear relationship (r2=0.9939) between Fluc activity and cell number confirms that the mES cells were successfully labeled with DiR dye. Fig.2C shows the result of trypan blue staining, which suggests that the DiR(+) mES cells had good viability similar with the DiR(-) mES cells. Fig.2D shows that DiR-mES cells grew very well after being labeled with DiR. These data highly suggest that DiR-mES cells had good cellular bioactivity and were suitable for in vivo imaging and tracking.
In vivo fluorescence imaging of DiR-mES cells in gastric cancer mouse models
In order to assess the biodistribution of mES cells in bioluminescence gastric cancer nude mouse models. DiR-GFP-mES cells were intravenously injected into gastric cancer mouse models as test group. In control group, gastric cancer mouse models were injected with DiR dye. The distribution of DiR-mES cells in gastric cancer nude mouse models was monitored within 24 h by IVIS imaging system at lateral, supine and prone positions. As shown in Fig.3, fluorescent signals in subcutaneous tumor tissues in test group were clearly differentiated from the surrounding tissues at 10 min post-injection at lateral position. Fluorescent signals in tumor tissues gradually increased, reaching a maximum value at 2 h post-injection. The higher tissue to background ratio (TBR) value suggests that the DiR-mES cells preferentially accumulated in tumor tissues and achieved their maximum value at 2 h post-injection. Conversely, no fluorescent signals could be observed in the tumor tissues in control group. In addition, the in vivo imaging of the mouse models was performed at supine position and prone position as shown in Supplementary Material: Fig.S1. These results show that DiR-mES cells mainly located in the tumor tissues, and very few DiR-mES cells located in the liver, lung and kidney organs, similar to several reports [24-26].
Fig 1
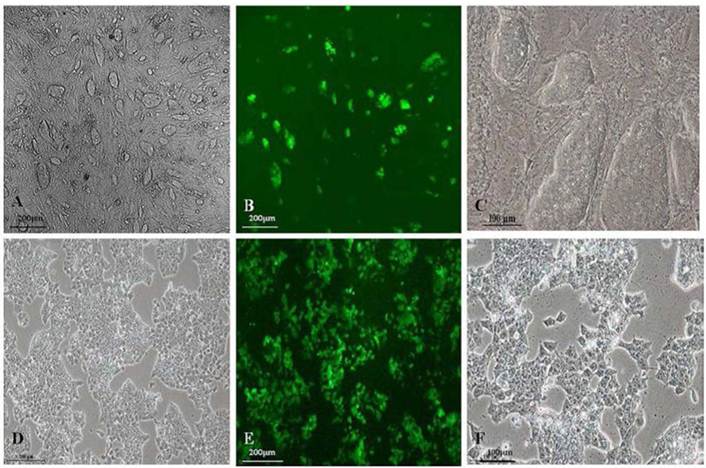
Fluorescence microscope images of the morphology of GFP-SV129 mES cells.(A) GFP-SV129 mES cells exhibited typical large colonies and clearly boundaries with feeder cells. (B) mES cells expressed strong green fluorescence. (C) Magnification of mES cells cultured on feeder cells. (D) Feeder-free culture of mES cells grew in monolayer. (E) Feeder-free culture of mES cells exhibited green fluorescence. (F) Magnification of feeder-free culture of mES cells.
Fig 2
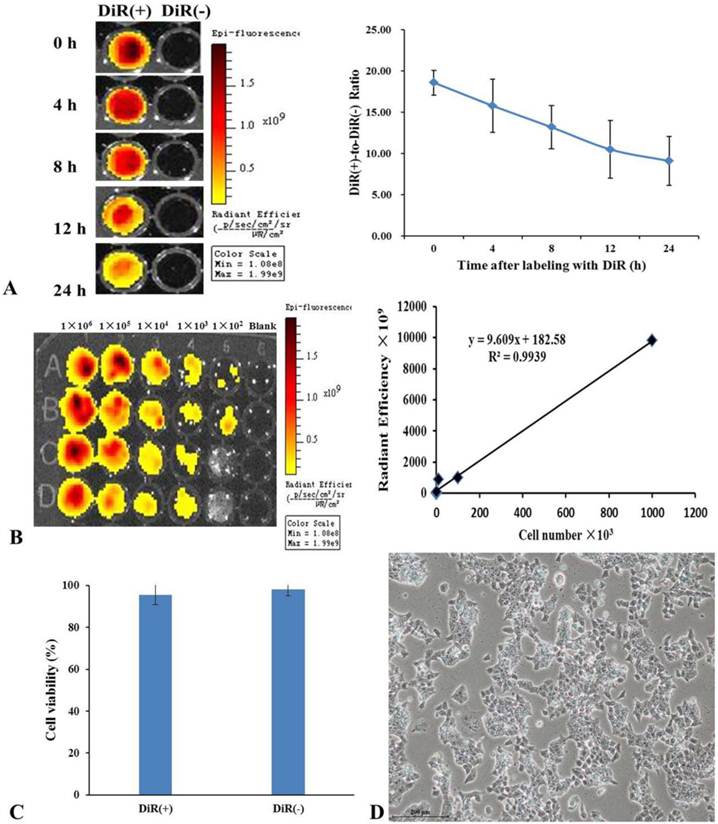
(A) DiR-labeled mES cells were subjected to fluorescent imaging at different exposure time, the ratio of DiR-labeled mES cells to unlabeled mES cells shows that the DiR-mES cells had strong fluorescent signals within 24 h. (B) The quantitative analysis of fluorescence of DiR(+) cells showed a linear relationship (r2=0.9939) between Fluc activity and cell numbers. (C) Trypan blue staining shows that mES cells had good cell viability after being labeled with DiR. (D) mES cells grew very well after being labeled with DiR 24 h.
Fig 3
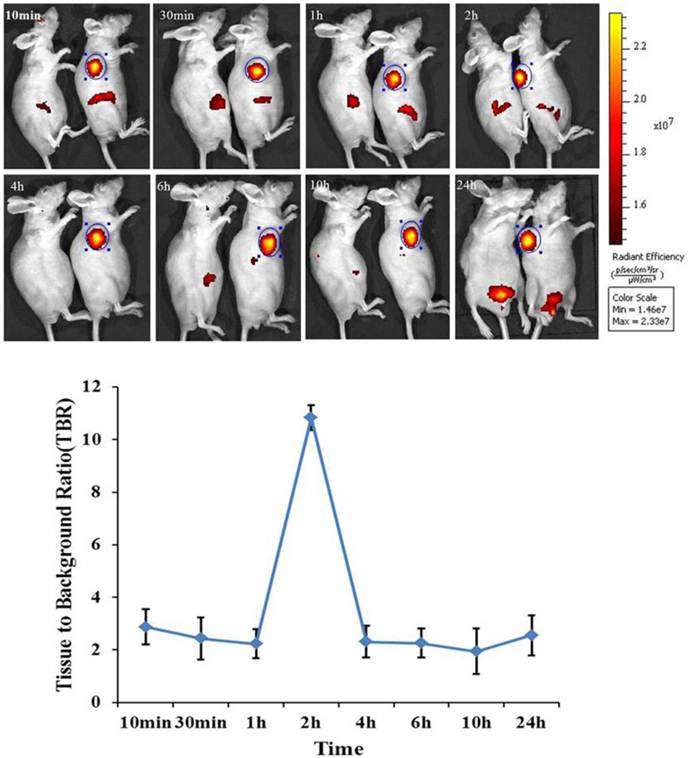
In vivo fluorescence images of gastric cancer mouse models at 10 min, 30 min, 1 h, 2 h, 4 h, 6 h, 10 h, 24 h post-injection DiR-mES cells at lateral position. It shows that tumor tissues had strong fluorescence signals after injection DiR-mES cells as shown in blue circles. The tissue to background ratio (TBR) values show that DiR-mES cells accumulated in the tumor tissues and reached peak value at 2 h post-injection.
In order to track the distribution of DiR-mES cells in gastric cancer mouse models, the gastric cancer mouse models were performed bioluminescence and fluorescence imaging at 6 h post-injection. Fig.4A shows the NIR fluorescence imaging of the gastric cancer mouse models. Fig.4B shows the bioluminescence imaging of the gastric cancer mouse models. Fig.4C is a combination image of bioluminescence and fluorescence imaging. These results fully confirm that DiR-mES cells could migrate to the tumor tissues precisely. As shown in Fig.4D, the ex vivo fluorescence imaging of major tissues in test and control groups shows that DiR-mES cells mainly existed in the tumor tissue of the test group. No obvious fluorescent signal were detected in the spleen and heart tissues of the test and control group, and weak fluorescent signals were found in the lung, kidney and liver tissues of the test group. The reason for fluorescent signals found in liver tissue may be as following: DiR-mES cells were cleared from circulation by the liver and trapped within capillary beds in the liver [27]. As shown in Fig.4E, the radiant efficiency of tumor tissue in test group was the highest among all the tissues in the test and control groups, which further confirm that DiR-mES cells could preferentially accumulate in the tumor tissue and efficiently target and identify gastric cancer cells in gastric cancer mouse models.
Pathological analysis of tumor tissues and important organs
Pathological evaluation of excised major tissues including the liver, lung, spleen, kidney, heart, and tumor was performed. SSEA-1 is a specific, highly expressed surface antigen of mES cells, so the tumor tissues must express SSEA-1 once the mES cells migrated into the tumor tissues. As shown in Fig.5A, GFP and SSEA-1 fluorescent signals were significant observed from the ultra-thin tumor tissue slices of the test group after performed immunofluorescence staining, and green fluorescence signals were consistent with fluorescent signals in the tumor tissues of the test group, no GFP and immunofluorescent signals were found in the tumor tissues of the control group. Furthermore, as shown in Supplementary Material: Fig.S2, no fluorescent signal was observed in the liver, lung, spleen, heart and kidney tissues of the test group. These results fully suggest that mES cells could specifically target gastric cancer cells in vivo.
Western blot analysis of gastric cancer tissues
The expression of SSEA-1 in excised tumor tissues was investigated using Western blotting. The β-actin protein was used as a reference. Fig.5B shows that SSEA-1 was highly expressed in the tumor tissues of the test group, while it did not express in the tumor tissues of the control group. This result suggests that mES cells mainly located in the tumor tissues in vivo.
Fig 4
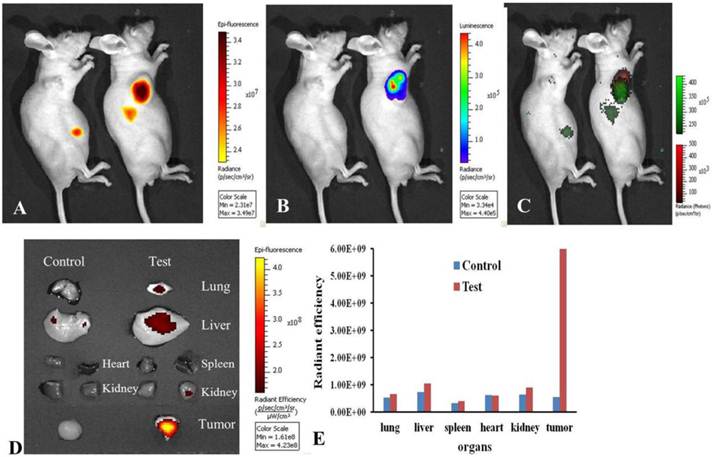
Optical imaging of DiR-mES cells in vivo/ex vivo. (A) Fluorescence imaging of gastric cancer mouse models at 6 h post-injection at lateral position; (B) Bioluminescence imaging of gastric cancer mouse models at 6 h post-injection at lateral position; (C) Combination image of fluorescence and bioluminescence imaging shows that DiR-mES cells could target the tumor tissues exactly. (D)Ex vivo fluorescent image of the dissected organs and tumors in the control group and test group at 24 h post-injection. (E)Radiant efficiency of the tissues in the test group and control group.
Fig 5
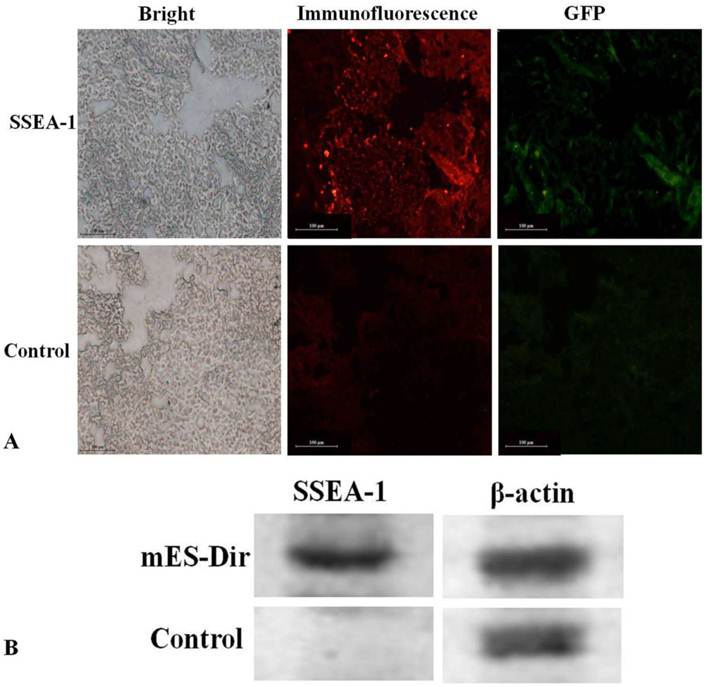
(A) Immunofluorescence analysis of the tumor tissues. (B) Western blot analysis of the tumor tissues.
Possible mechanism of the migration of mES cells to gastric cancer cells
Tumor-homing is a complex, multistep process in which many cells move from distant locations to a tumor site. Homing mES cells may be activated by specific signals, and they can travel through the circulation, extravasate from vessels, migrate, and undergo phenotypic changes when it finally reaches the tumor site. The mechanism might be similar to the metastatic cascade [28]. So far, the process of tumor-homing associated with mES cells is still not clarified. The term “tumor-homing” indicates any action in which cells travel from a distant location to a tumor site, indicating an active filopodia-based motion through a tumor or surrounding local microenvironment based on local chemoattractants might be involved [29-36]. For example, CXCR4-CXCL12 loop was in charge of metastasis of the breast cancer [37]. The interaction between CXCL12 and CXCR4 has been implicated in the bone metastasis in prostate cancer [38]. The CXCL12/CXCR4 axis has been shown to be involved in the metastasis of non-small-cell lung cancer cells [39]. Thus, we speculate that CXCL12-CXCR4 loop may be involved in the course of mES cells migrating to the gastric cancer cells in vivo.
As shown in Fig.6, flow cytometer analysis results show that the positive rate of CXCR4 expression was 9.46 % in mES cells. ELISA results show that amounts of CXCL12 in the culture supernatant of MFC cells were 7.2±0.7 ng/mL. Cell and chemoattractant dose-response curves show that CXCL12 induced chemotaxis of mES cells in a dose-dependent, bell-shaped manner with maximum chemotactic response seen at 400 ng/mL of CXCL12. According to these results, we suggested a possible mechanism of mES cells migrating to gastric cancer cells in vivo. DiR-labeled mES cells enter the tail vein vessels, and then rapidly circulate into lung vessels. From there, they enter into artery vessels, circulating throughout the mouse body. Some mES cells enter the liver and are captured and endocytosed by the phagocyte, finally are trapped in the liver. High amounts of CXCL12 were found in the surrounding of the tumor tissue, providing a strong chemotactic signal. Since mES cells positive expressed CXCR4 receptor, they are very sensitive to CXCL12 factor secreted by gastric cancer tissue, which triggers the rapid mES cells migration toward gastric cancer tissues. In the course of migration of mES cells to gastric cancer tissues, it is possible that some other chemokine factors are also involved, therefore, a comprehensive study evaluating other potential mechanisms is under way.
Stem cell therapy is a promising method for cancer treatment [40-42]. Especially in 2004, there is one research reported that when the human embryonic stem (hES) cells were transfected with an anticancer molecular named “TRAIL”, the transfected hES cells could migrate to the brain tumor site and significant prevent the tumor growth [43]. The targeting ability is the key for stem cell therapy of tumors. In this research, we clearly observed that mES cells have good ability of targeting gastric cancer. This discovery lays foundation for diagnosing early gastric cancer with embryonic stem cells.
In this study, our results indicate that DiR-labeled mES cells could target and recognize cancer cells in vivo. In addition to gastric cancer mouse model, nude mouse models with lung cancer, liver cancer, and prostate cancer were also prepared. It is found that DiR-labeled mES cells could target and recognize those different cancer cells in vivo as well (data not shown). However, the detailed molecular mechanism of tumor targeting still requires further investigation.
Fig 6
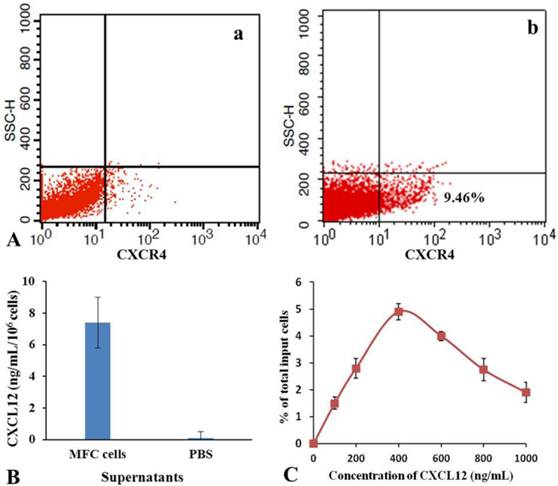
Analysis of chemotaxis. (A) Expression of CXCR4 in isotype control(a) and mES cells (b) were determined by flow cytometry. (B) Secretion of CXCL12 in MFC cells was measured by enzyme-linked immunosorbent assay. (C) Cell and chemoattractant dose-response curves.
Conclusions
In conclusion, this is the first report showing that DiR-labeled mES cells can selectively migrate to gastric cancer tissues in vivo and can be used for targeted NIR imaging of gastric cancer cells in vivo. The ligand-receptor combinations of CXCL12 and CXCR4 may be responsible for the migration of mES cells to the gastric cancer cells. Although the detailed mechanism still requires further investigation, we believe that this phenomenon may be general in other tumor models. Therefore, it has great potential in imaging detection of early and metastatic cancer cells in the near future.
Supplementary Material
Fig.S1 - S2.
Acknowledgements
This work is supported by National Key Basic Research Program (973 Project) (No. 2010CB933901), Important National Science & Technology Specific Projects (2009ZX10004-311), National Natural Scientific Fund (No. 31100717), New Century Excellent Talent of Ministry of Education of China (NCET-08-0350), Special project for nanotechnology from Shanghai (1052nm04100), Shanghai Jiao Tong University Innovation Fund for Postgraduates (No.AE340201), Shanghai Jiao Tong University Investment Fund for Excellent Doctoral Dissertation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1981;78:7634-8
2. Reubinoff BE, Pera MF, Fong CY. et al. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nature Biotechnology. 2000;18:399-404
3. Itskovitz-Eldor J, Schuldiner M, Karsenti D. et al. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol Med. 2000;6:88-95
4. Hoehn M, Kustermann E, Blunk J. et al. Monitoring of implanted stem cell migration in vivo: a highly resolved in vivo magnetic resonance imaging investigation of experimental stroke in rat. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:16267-72
5. Lapidot T. Mechanism of human stem cell migration and repopulation of NOD/SCID and B2mnull NOD/SCID mice. Annals of the New York Academy of Sciences. 2001;938:83-95
6. Forte G, Minieri M, Cossa P. et al. Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells. 2006;24:23-33
7. Prockop DJ, Gregory CA, Spees JL. One strategy for cell and gene therapy: harnessing the power of adult stem cells to repair tissues. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:11917-23
8. Cavazzana-Calvo M, Hacein-Bey S, Basile GS. et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669-72
9. Krause DS, Theise ND, Collector MI. et al. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369-77
10. Korbling M, Estrov Z. Adult stem cells for tissue repair-a new therapeutic concept. N Engl J Med. 2003;349:570-82
11. Xu AG, Li SG, Liu JH, Gan AH. Function of apoptosis and expression of the proteins Bcl-2, p53 and C-myc in the development of gastric cancer. World J Gastroentero. 2001;7:403-6
12. Pisani P, Parkin DM, Bray F. et al. Estimates of the worldwide mortality from 25 cancers in 1990. International Journal of Cancer. 1999;83:18-29
13. Roder DM. The epidemiology of gastric cancer. Gastric cancer. 2002;5:5-11
14. Cui D, Zhang L, Yan X. et al. A microarray-based gastric carcinoma prewarning system. World J Gastroenterol. 2005;11:1273-82
15. Li Z, Huang P, He R. et al. Aptamer-conjugated dendrimer-modified quantum dots for cancer cell targeting and imaging. Materials Letters. 2010;64:375-8
16. Li Z, Huang P, Zhang X. et al. RGD-conjugated dendrimer-modified gold nanorods for in vivo tumor targeting and photothermal therapy. Molecular Pharmaceutics. 2009;7:94-104
17. Huang P, Li Z, Lin J. et al. Photosensitizer-conjugated magnetic nanoparticles for in vivo simultaneous magnetofluorescent imaging and targeting therapy. Biomaterials. 2011;32:3447-58
18. Wang K, Ruan J, Qian Q. et al. BRCAA1 monoclonal antibody conjugated fluorescent magnetic nanoparticles for in vivo targeted magnetofluorescent imaging of gastric cancer. Journal of Nanobiotechnology. 2011;9:23
19. Kovar JL, Simpson MA, Schutz-Geschwender A. et al. A systematic approach to the development of fluorescent contrast agents for optical imaging of mouse cancer models. Analytical Biochemistry. 2007;367:1-12
20. Jenkins DE, Oei Y, Hornig YS. et al. Bioluminescent imaging (BLI) to improve and refine traditional murine models of tumor growth and metastasis. Clinical and Experimental Metastasis. 2003;20:733-44
21. Weissleder R. Molecular imaging in cancer. Science. 2006;312:1168-71
22. Herschman HR. Molecular imaging: looking at problems, seeing solutions. Science. 2003;302:605-8
23. Frangioni JV. In vivo near-infrared fluorescence imaging. Current Opinion in Chemical Biology. 2003;7:626-34
24. Vilalta M, Degano IR, Bago J. et al. Biodistribution, Long-term Survival, and Safety of Human Adipose Tissue-derived Mesenchymal Stem Cells Transplanted in Nude Mice by High Sensitivity Non-invasive Bioluminescence Imaging. Stem Cells Dev. 2008;17:993-1003
25. Choi KS, Ahn SY, Kim TS. et al. Characterization and Biodistribution of Human Mesenchymal Stem Cells Transduced with Lentiviral-mediated BMP2. Arch Pharm Res. 2011;34:599-606
26. Detante O, Moisan A, Dimastromatteo J. et al. Intravenous Administration of 99mTc-HMPAO-Labeled Human Mesenchymal Stem Cells After Stroke: In Vivo Imaging and Biodistribution. Cell Transplant. 2009;18:1369-79
27. Ilium L, Davis SS, Wilson CG. et al. Blood clearance and organ deposition of intravenously administered colloidal particles. The effects of particle size, nature and shape. International Journal of Pharmaceutics. 1982;12:135-46
28. Reagan MR, Kaplan DL. Concise Review: Mesenchymal Stem Cell Tumor-Homing: Detection Methods in Disease Model Systems. Stem Cells. 2011;29:920-7
29. Suda Y, Suzuki M, Ikawa Y. et al. Mouse embryonic stem cells exhibit indefinite proliferative potential. J Cell Physiol. 1987;133:197-201
30. Hirofumi S, Yuzo K, Norio N. A mouse embryonic stem cell line showing pluripotency of differentiation in early embryos and ubiquitous β-galactosidase expression. Cell Differentiation and Development. 1990;29:181-6
31. Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nature Genetics. 2000;24:372-6
32. Raman D, Baugher PJ, Thu YM, Richmond A. Role of chemokines in tumor growth. Cancer Letters. 2007;256:137-65
33. Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Letters. 2008;267:226-44
34. Molyneaux KA, Zinszner H, Kunwar PS. et al. The chemokine SDF1/CXCL12 and its receptor CXCR4 regulate mouse germ cell migration and survival. Development. 2003;130:4279-86
35. Son BR, Marquez-Curtis LA, Kucia M. et al. Migration of bone marrow and cord blood mesenchymal stem cells in vitro is regulated by stromal-derived factor-1-cxcr4 and hepatocyte growth factor-c-met axes and involves matrix metalloproteinases. Stem Cells. 2006;24:1254-64
36. Guo Y, Hangoc G, Bian H. et al. SDF-1/CXCL12 enhances survival and chemotaxis of murine embryonic stem cells and production of primitive and definitive hematopoietic progenitor cells. Stem Cells. 2005;23:1324-32
37. Muller A, Homey B, Soto H. et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50-6
38. Taichman RS, Cooper C, Keller ET. et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Research. 2002;62:1832
39. Phillips RJ, Burdick MD, Lutz M. et al. The Stromal Derived Factor-1/CXCL12-CXC Chemokine Receptor 4 Biological Axis in Non-Small Cell Lung Cancer Metastases. American Journal of Respiratory and Critical Care Medicine. 2003;167:1676-86
40. Strauer BE, Kornowski R. Mini-Review: Expert Opinions. Circulation. 2003;107:929-34
41. Yates F, Daley G. Progress and prospects: gene transfer into embryonic stem cells. Gene Therapy. 2006;13:1431-9
42. Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nature Reviews Cancer. 2002;2:750-63
43. Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371-8
Author contact
![]() Corresponding author: Daxiang Cui, Department of Bio-Nano Science and Engineering, Research Institute of Micro/Nano Science and Technology, Shanghai Jiao Tong University, 800Dongchuan Road, Shanghai200240, P. R. China. Tel: +86-21-34206375. Fax: +86-21-34206886. E-mail: dxcuiedu.cn.
Corresponding author: Daxiang Cui, Department of Bio-Nano Science and Engineering, Research Institute of Micro/Nano Science and Technology, Shanghai Jiao Tong University, 800Dongchuan Road, Shanghai200240, P. R. China. Tel: +86-21-34206375. Fax: +86-21-34206886. E-mail: dxcuiedu.cn.
Citation styles
APA
Ruan, J., Song, H., Li, C., Bao, C., Fu, H., Wang, K., Ni, J., Cui, D. (2012). DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells. Theranostics, 2(6), 618-628. https://doi.org/10.7150/thno.4561.
ACS
Ruan, J.; Song, H.; Li, C.; Bao, C.; Fu, H.; Wang, K.; Ni, J.; Cui, D. DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells. Theranostics 2012, 2 (6), 618-628. DOI: 10.7150/thno.4561.
NLM
Ruan J, Song H, Li C, Bao C, Fu H, Wang K, Ni J, Cui D. DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells. Theranostics 2012; 2(6):618-628. doi:10.7150/thno.4561. https://www.thno.org/v02p0618.htm
CSE
Ruan J, Song H, Li C, Bao C, Fu H, Wang K, Ni J, Cui D. 2012. DiR-labeled Embryonic Stem Cells for Targeted Imaging of in vivo Gastric Cancer Cells. Theranostics. 2(6):618-628.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.