Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Reporter genes commonly used for...
Marking immune cell populations...
Monitoring immune cell function
Challenges ahead
Conclusion
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2012; 2(4):355-362. doi:10.7150/thno.3903 This issue Cite
Review
Reporter Gene Imaging of Immune Responses to Cancer: Progress and Challenges
Purnima Dubey ![]()
Departments of Pathology-Tumor Biology, Cancer Biology, Microbiology & Immunology, Comprehensive Cancer Center, Wake Forest School of Medicine, Winston Salem, NC 27157-1092, USA
Received 2011-12-2; Accepted 2012-2-8; Published 2012-4-4
Abstract
Immune responses to cancer are dynamic processes which take place through the concerted activity of innate and adaptive cell populations. In order to fully understand the efficacy of immune therapies for cancer, it is critical to understand how the treatment modulates the function of each cell type involved in the anti-tumor immune response. Molecular imaging is a versatile method for longitudinal studies of cellular localization and function. The development of reporter genes for tracking cell movement and function was a powerful addition to the immunologist's toolbox. This review will highlight the advances and challenges in the use of reporter gene imaging to track immune cell localization and function in cancer.
Keywords: reporter gene imaging, immune responses, cancer
Introduction
The immune system has the capacity to recognize and destroy malignant cells, a process which is thought to occur regularly during the mammalian lifespan. Immune surveillance of cancer is mediated by both innate and adaptive cells [1], whose interaction determines whether the outcome will be progressive tumor growth or tumor rejection. Innate immune cells comprise myeloid-derived cells such as natural killer cells, and phagocytes such as dendritic cells, macrophages, and neutrophils, which engulf and remove pathogens from tissues and peripheral blood. Immune surveillance of nascent transformed cells includes destruction of tumor cells by these professional phagocytes. In addition, these innate immune cells can modulate the function of adaptive immune cells throughout tumor progression, by differential production of cytokines and chemokines.
Mature B and T cells, the broad categories of adaptive immune cells, mature in the bone marrow and thymus, respectively. Antibodies are produced by B cells in response to antigens presented by dendritic cells and macrophages. Thymus-derived T cells include CD8+ cytotoxic T cells with direct lytic capacity, CD4+ helper T cells which shape the immune response by production of cytokines, and CD4+FoxP3+ regulatory T cells which modulate the function of both CD8+ and CD4+ T cells. Antigens presented by innate immune cells can activate all three types of T cells, which then either augment or inhibit tumor growth [2].
Thus, the interaction between innate and adaptive immune cells within the tumor microenvironment modulates cancer development and progression. Currently, this dynamic process is largely evaluated with snapshots in time and space. Immune cells harvested from tumor biopsies, excised tumor tissue, or peripheral blood are analyzed by immunohistochemistry or flow cytometry to determine the number and functional status of cells within the tumor and host. Although highly informative, these analyses cannot evaluate changes in cell numbers or functional status within the same living subject. Functional changes over time are, thus, evaluated using serial measurements of multiple experimental animals in pre-clinical models, and multiple biopsies in patients when available.
Non-invasive molecular imaging is uniquely poised to provide dynamic spatial and temporal information on the progress of an immune response in living subjects [3, 4]. Approaches to track immune cells have utilized both direct and indirect labeling methods. Direct labeling approaches for MRI and PET have been used to image both adaptive and innate immune cells, and have been reviewed elsewhere [5, 6]. However, the label is diluted when the cells divide, thus providing only a limited time frame for tracking cell movement in vivo. In contrast, indirect labeling via genetic introduction of reporter genes permanently marks cells of interest, permitting long-term tracking of immune responses. In addition, false positives are avoided, since dead cells do not produce an enzyme-substrate reaction and, thus, do not emit a signal. This review will highlight the progress made in tracking immune cell localization and function in cancer using non-invasive whole-body reporter gene imaging, and discuss the limitations of current approaches. Cell-cell interactions in the immune system have been studied using fluorescent reporter genes and intravital microscopy techniques, and have been reviewed elsewhere [7, 8].
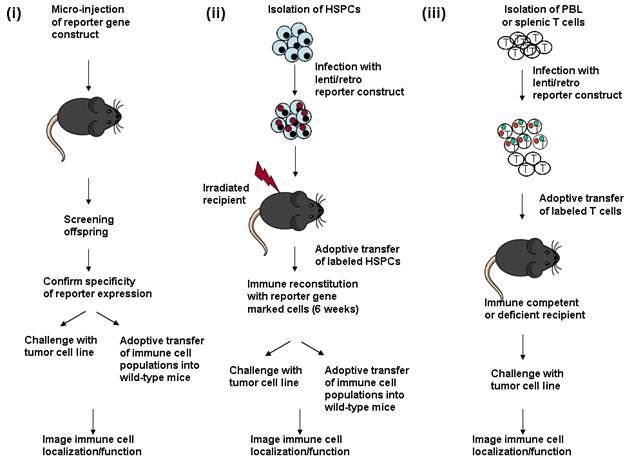
Introduction of imaging reporter genes into immune cell populations to follow an anti-tumor response. (i) Micro-injection of reporter gene DNA into fertilized oocytes to create transgenic mice. Genomic tail DNA is screened for presence of the transgene. Expression of the reporter gene is then determined by analysis of bone marrow, spleen or thymus cell populations, generally by flow cytometry. If a cell or tissue-restricted promoter is used, specificity is confirmed by analysis of reporter gene expression in individual cell types. The transgenic offspring can either be directly challenged with a tumor cell line to follow immune cell localization during tumor growth. Alternately, different immune cell subsets expressing the reporter gene can be adoptively transferred into wild-type mice which are then challenged with the tumor to follow immune cell localization/function. (ii) HSPCs (hematopoietic stem progenitor cells) are isolated from the bone marrow of wild-type mice, and cultured briefly in vitro for infection with a lentivirus or retrovirus expressing a reporter gene construct. Recipient animals are lethally irradiated to ablate the endogenous immune system, and then injected with the infected bone marrow cells. The immune system is reconstituted within 6 weeks after transfer of marked bone marrow, and the reconstitution process can be followed by bioluminescent or radioisotopic imaging. The reconstituted animals are either directly challenged with a tumor cell line, to follow cell localization and function, or the marked immune cell populations can be serially transferred into secondary recipient animals. (iii) Immune cell populations (generally T cells) are isolated from peripheral blood (PBL) or spleen, stimulated to proliferate in vitro and infected with a lentivirus or retrovirus expressing the reporter construct, following short term culture. The labeled cell population is then injected into immune competent or immune deficient mice to study their localization and function at the site of a tumor.
Reporter genes commonly used for imaging immune cells
Reporter genes have been developed for both radioisotopic and optical non-invasive imaging techniques. For pre-clinical imaging, optical methods provide greater sensitivity of signal, while radioisotopic techniques are applicable for imaging in patients. The strengths and limitations of these two techniques have been recently reviewed [5, 9, 10] .
Several reporter genes for single- and multi-modality imaging have been developed [11]. The most widely used bioluminescent reporter gene is Firefly luciferase, derived from the firefly Phontinus pyralis [12]. Its substrate, luciferin, requires oxygen and ATP, and emits a strong signal particularly close to the body surface. Codon-optimized versions of this enzyme can detect as few as 3 T cells localized subcutaneously in a mouse [13].
Renilla luciferase, derived from the sea pansy Renilla reniformis, uses the substrate coelenterazine [13]. Mutated versions of Renilla luciferase with greater stability and signal strength have also been created [14]. Although the emission spectra of Firefly and Renilla luciferase overlap, their kinetics of emission are very different, permitting dual imaging of two signals in the same animal, nearly simultaneously. Gaussia luciferase [15] also uses coelenterazine as its substrate and can be either secreted, or membrane tethered [16]. Secreted Gaussia luciferase in the supernatant can be quantified to determine activity in the cell population, while the number of cells expressing the membrane-tethered variant can be analyzed by flow cytometry.
For radioisotopic molecular imaging, the viral thymidine kinase gene remains the reporter gene of choice. The most widely used is Herpes Simplex Virus Thymidine Kinase (HSV-TK) [4], and both wild-type and mutated versions of this enzyme are used for PET imaging. Both 18F-labeled and 124I-labeled substrates have been developed which are phosphorylated by mutant or wild-type HSV-TK [17]. Recently, a mutant human thymidine kinase 2 gene was created which phosphorylates a substrate for viral thymidine kinase [18, 19]. The human thymidine kinase as a PET reporter gene is advantagenous since it is non-immunogenic in humans, thus alleviating the potential problem of rejection of cells transduced with the viral thymidine kinase.
Fluorescent reporter genes incorporated in imaging constructs include GFP or red fluorescent protein (RFP). Generally, these genes are used for quantitation of infected cells, but not non-invasive imaging of immune cells, since sensitivity and resolution of these reporters is lower than both bioluminescent and radioisotopic imaging reporters.
Several groups have developed multipurpose reporters, which incorporate a fluorescent, bioluminescent and radioisotopic reporter in the same construct [20-22]. This approach allows quantitation of the number of reporter gene-expressing cells using the fluorescent reporter, and sequential bioluminescent and radioisotopic imaging of the same animal. Consequently, the sensitivity of bioluminescence is combined with the spatial resolution of PET imaging, providing a comprehensive picture of the immune response. Multi-modality reporter constructs are created by making direct fusions of all three elements separated by flexible linkers, or expressing the individual elements on the same cistron, separated by internal ribosomal entry site elements or ribosomal slippage sites [23]. A possible limitation of these fusion genes may be attenuated signal from one or more of the components, compared with expression of each reporter gene alone. Thus, the advantage of multimodality imaging should be weighed together with the potential loss of signal strength. Both single modality and multimodality fusion reporters have been used to image immune cell subsets.
Marking immune cell populations with reporter genes
In order to dissect the interaction of different cell types in vivo, expression of a reporter gene must be driven behind a cell type-specific promoter. One means of obtaining restricted reporter gene expression is creation of transgenic mice. One of the earliest reports which studied T cell localization by reporter gene imaging used the T-cell specific human CD2 promoter to drive expression of the Firefly luciferase reporter gene in all T cells. This transgenic mouse strain was crossed with CD8+ [24] or CD4+ [25] T cell receptor transgenic mice, creating double transgenic animals with expression of luciferase in antigen-specific CD8+ T cells or CD4+ T cells, respectively. This powerful approach permits longitudinal tracking of antigen-specific T cells in vivo, and has been used to follow responses to model antigens. The kinetics of T cell localization to transplantable tumors can be directly evaluated by challenging the transgenic mice with transplantable tumors expressing the model antigen. The limitation of this approach, however, is the need to create two different transgenic strains of mice, which can each track only one model antigen.
Control of T cell function by Tregs prevents expansion and effector activity of CD8+ and CD4+ T cells which respond to cancers. Modulation of Tregs has been widely used as a means of augmenting anti-tumor immune responses in animal models, and also in the clinic [26, 27]. The ability to visualize Tregs in vivo would provide an important means of tracking the effects of Tregs on vaccinations or other therapies intended to increase effector T cell numbers and function. Several different strains of mice have been created using knockin or knockout technology (reviewed in [28]) with fluorescent and/or bioluminescent reporters expressed alone under the control of FoxP3 regulatory elements, or driving a fusion or bicistronic protein which includes a reporter gene together with the FoxP3 coding sequence. These strains have been used to study normal Treg function, but so far have not been used to study responses to tumors.
Dendritic cells, macrophages and neutrophils in cancers all have context-dependent functions of T cell activation or inhibition, and the interaction of these cells with T cell subsets modulates tumor progression [2]. A transgenic mouse was created driving expression of GFP behind the Lysozyme M (LysM) promoter [29], and was used to track macrophage localization in infection. Intra-tumoral macrophages can augment effector cell function in cancer, depending on the cytokines, chemokines, or activation products produced by the tumor or the macrophages themselves. In vivo visualization of the waxing and waning of this cell population in tumors would dissect another important arm of the antitumor response.
Dendritic cells (DCs), the professional antigen presenting cells of the immune system, are also studied as a potential cancer therapy. Adoptive transfer of this cell population after modification in vitro can activate effector immune responses against tumors and modulate autoimmune responses. DCs purified from a Firefly luciferase-expressing transgenic mouse showed the distribution pattern of these cells in the recipient host, an important criterion for determining the ability of transferred cells to induce an immune response in the target tissue [30].
An approach which bypasses creation of subset-specific transgenic reporter mice is adoptive transfer of individual cell populations isolated from an animal expressing luciferase behind a ubiquitous constitutive promoter such as β-actin [31]. The contribution of the labeled cell population to responses in an immune competent animal can then be studied. However, the endogenous population is often ablated prior to adoptive transfer, perturbing the immune system.
Adoptive transfer of cell populations purified from autologous hematopoietic stem cells or peripheral blood, are used in the clinical setting as a treatment for cancer [32]. Lentivirus and retroviral vectors have been used to transduce stem cells [33] and primary isolated cell populations [34-36] with imaging reporter genes. Constitutive expression in all infected cells results from LTR-driven expression in retroviruses. This approach was used to visualize the localization of CD8+ T cells to a virally induced tumor [37]. Splenocytes of mice which had previously rejected a challenge of the Murine Sarcoma Virus/Murine Leukemia Virus were isolated and infected with a retroviral construct expressing a HSV-TK-GFP fusion protein. GFP+CD8+ T cells were isolated and adoptively transferred into immune deficient mice bearing the virally induced tumor. The localization of these memory CD8+ T cells was monitored by PET imaging using 18F-FHBG as the substrate. This study demonstrated that serial PET imaging could be used to monitor T cell localization to a growing tumor. In a conceptually similar study, a human T cell line was retrovirally transduced with the HSV-TK PET reporter gene and localization to an Epstein Barr Virus-expressing tumor was demonstrated using 124I-FIAU as the substrate [38]. This approach has also been used to demonstrate the homing and survival in vivo of bone marrow-derived dendritic cells derived and transduced with a GFP and Firefly luciferase fusion protein ex vivo [36].
Ubiquitous or cell-type specific expression can be achieved in lentiviruses, where reporter gene expression is driven by internal tissue- or cell-specific promoters. Control elements from the human CD4 promoter were used to drive CD4 T cell-restricted expression of Green Fluorescent Protein (GFP) in a lentiviral construct [39]. This reporter construct was slightly leaky, with GFP expression observed in a fraction of other T cells and B cells [40]. Nevertheless, the largely CD4-restricted expression allowed flow cytometric monitoring of these cells following adoptive transfer of the labeled population, and would be useful for tracking the contribution of CD4+ T cells to an anti-tumor immune response in vivo. However, this strategy may have limited application since genomic control regions may be as far as 100 kb removed from the minimal promoter [41], and placing regulatory elements outside their genomic context often abrogates specificity.
A potential adverse effect of retroviral or lentiviral infection is mutation resulting from the site of insertion of the viral genome, which may activate a proto-oncogene or inactivate a tumor suppressor gene, leading to transformation of the transduced cell population [42-44]. In addition, immune function of the infected cell may be altered by viral infection.
Adoptive transfer of reporter gene-labeled cells into immune competent mice poses the potential risk of immune elimination of the transferred cells which recognize the reporter gene as a foreign antigen, and has been observed in some instances [45-48]. Mutated versions of human thymidine kinase 2 have been reported which also react with PET reporter probes developed for viral thymidine kinase [18, 19]. The use of human thymidine kinase would prevent an immune response against the reporter gene, making this type of reporter more useful for clinical imaging.
An alternative approach is to introduce the reporter gene into hematopoietic stem cells. An intact naive immune system with reporter gene labeled cell populations can be created by infection of hematopoietic stem/progenitor cells with lentiviral and retroviral constructs. Adoptive transfer of the infected progenitor population into lethally irradiated recipients creates an immune system with bone marrow-derived myeloid and lymphoid cells marked with imaging reporter genes. The reporter gene is now a “self-antigen”, and marked cells will not be recognized and eliminated by the host immune system.
This approach has been used to introduce both bioluminescent and radioisotopic reporter genes into immune cell components using ubiquitously expressed promoters [33], and was used to monitor primary immune responses to experimental cancers [49]. The time course and magnitude of memory responses to tumor antigens can also be followed with this system. This strategy is faster than creation of a transgenic mouse, and permits tracking of immune responses in any genetic background. The limitations for subset specificity, however, remain the same as discussed above. Furthermore, hematopoietic stem cells and primary lymphocytes are relatively resistant to infection, and are transduced with low efficiency, despite advances in viral gene transfer methodology. Thus, only a subset of the responding cell population is labeled with the imaging reporter. Nevertheless, the extent of labeling is sufficient to monitor cell localization to the tumor or site of infection.
Monitoring immune cell function
Activation of immune cells which respond to pathogens or cancers is a dynamic process. For example, T cells require three signals in order to become fully functional [50]. Signal 1 is engagement of the T cell receptor by interaction with the MHC molecule presenting the antigen. Subsequently, interaction of co-stimulatory receptors on T cells with their ligands on antigen-presenting cells delivers Signal 2. Secretion of cytokines which serve as growth factors is Signal 3, and results in T cell proliferation and effector function. The ability to visualize these activation steps would provide a more complete picture of the anti-tumor immune response and a better gauge of therapeutic efficacy. Translocation of the transcription factor NF-AT (Nuclear Factor of Activated T cells) to the nucleus rapidly follows ligation of the T cell receptor, and activates transcription of cytokine genes [51]. A lentivirus was created using the NF-AT DNA binding region, which controlled expression of the HSV-TK PET reporter gene. Infection of a T cell line with this lentivirus allowed visualization of Signal 1 after TCR triggering with anti-CD3 antibody [52]. Infection of murine splenocytes with a similar construct also allowed activation-dependent expression of luciferase, and visualization of activated T cells at the site of a tumor [5]. Subsequently, a lentivirus construct was created which permitted simultaneous visualization of T cell localization and NF-AT activation, using one reporter construct [35]. An insulator sequence was used to separate two promoters, which drove expression of two different luciferase reporter genes, one constitutive and the other activation-dependent. These constructs permit imaging of Signal 1 in T cell activation.
However, T cells which receive Signal 1, but not Signals 2 and 3 are anergic (unresponsive) and incapable of mounting effective responses [53]. Imaging NF-AT-dependent expression of reporter genes does not distinguish between anergic and fully responsive cells. CD8+ T cells which are fully functional produce cytotoxic products such as perforins and granzymes, which make holes in target cells. Recently, a lentivirus construct was reported which drove expression of Firefly luciferase behind the Granzyme B promoter. A transgenic CD8+ T cell line was transduced with this construct, which expressed luciferase after interaction with its cognate antigen, demonstrating that T cells with the capacity to lyse target cells can be detected using this reporter construct [54].
Challenges ahead
Studies to date have followed the localization and function of one cell type in an immune response. Since responses to cancers and pathogens involves the activity of several innate and adaptive immune cell types, the development of reporter gene constructs which allow tracking of multiple cell types simultaneously within the same animal will provide a more complete picture of therapeutic outcome. This can be accomplished by the use of two different imaging modalities sequentially, or by using two different bioluminescent reporters with different emisson kinetics, or two fluorescent reporters with non-overlapping emission spectra. The limitation, however, is the lack of cell-type specific promoters which can be incorporated in a viral vector. In addition, for clinical imaging, the use of reporter gene technology requires the development of safe and efficient gene transfer vectors.
Transduction with viral vectors is the most efficient method for introduction of reporter genes into hematopoietic stem/progenitor cells and immune cell types. However, the cells may be marked with as few as one viral copy per cell. Consequently, the reporter gene signal may be low, making it difficult to detect small numbers of responding immune cells. In addition, transduction of immune cells requires in vitro culture and stimulation with growth factors. Subsequent adoptive transfer into recipients requires depletion of at least some endogenous components, which alters the normal immune system, and is a limitation in animal studies. In the clinical setting, myeloablation is used as pre-conditioning prior to adoptive transfer of immune cells expanded in vitro [55], and transfer of reporter gene-marked immune cells would complement this protocol. This “passive immunotherapy” approach has been used to test the anti-tumor function of chimeric T cells and T cell receptor transgenic cells. In several instances the luciferase gene has been incorporated in the construct for bioluminescent imaging and the HSV-TK gene has been incorporated for PET imaging of the transferred T cells [56-60]. The incorporation of reporter constructs which are surrogates for T cell function will further enhance this technology.
Active immunotherapy attempts to generate an immune response against the cancer, by either blocking inhibitory receptors or cell populations, or generating T cell responses to antigens through vaccination of animals or human patients. In these protocols, reporter gene imaging has limited utility, since the goal is to observe the effect of immune manipulation on endogenous cell populations with the tumor or lymphoid organs. The identification of “image-able targets” within the intact immune system for which optical or radioactive tracers can be developed would circumvent this limitation. One such PET probe, [(18)F]FAC (1-[2'-deoxy-2'-[(18)F]fluoroarabinofuranosyl] cytosine), was used to visualize lymphoid organs and localized activation of anti-tumor T cell responses [61]. The development of other probes which may selectively image immune activation are currently underway [62].
Conclusion
The development of reporter gene imaging methods has provided tumor immunologists with powerful tools to study immune cell localization and function in vivo. Non-invasive longitudinal imaging of innate and adaptive immune subsets will reduce the number of experimental animals needed to achieve significant results, since the variation inherent in the analysis of different animals for several time points will be reduced. In preclinical models of therapy, the interaction of more than one immune cell subset can be studied nearly simultaneously. Non-invasive whole body imaging allows concurrent analysis of different anatomical locations, further accelerating evaluations of therapeutic efficacy. Thus, with the inclusion of reporter gene imaging in preclinical studies and clinical trials, novel immune therapies for cancer may be translated for use in patients at a more rapid pace.
Acknowledgements
This work was supported by American Cancer Society Research Scholar Grant 01-196-01. The author thanks Dr. Mark Willingham for critical reading of the manuscript.
Competing Interests
None.
References
1. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565-70
2. Shanker A, Marincola FM. Cooperativity of adaptive and innate immunity: implications for cancer therapy. Cancer Immunol Immunother. 2011;60:1061-74
3. Gross S, Moss BL, Piwnica-Worms D. Veni, vidi, vici: in vivo molecular imaging of immune response. Immunity. 2007;27:533-8
4. Massoud TF, Gambhir SS. Molecular imaging in living subjects: seeing fundamental biological processes in a new light. Genes Dev. 2003;17:545-80
5. Akins EJ, Dubey P. Noninvasive imaging of cell-mediated therapy for treatment of cancer. J Nucl Med. 2008;49(Suppl 2):180S-95S
6. Ottobrini L, Martelli C, Trabattoni DL, Clerici M, Lucignani G. In vivo imaging of immune cell trafficking in cancer. Eur J Nucl Med Mol Imaging. 2011;38:949-68
7. Celli S, Bousso P. Intravital two-photon imaging of T-cell priming and tolerance in the lymph node. Methods Mol Biol. 2007;380:355-63
8. Sumen C, Mempel TR, Mazo IB, von Andrian UH. Intravital microscopy: visualizing immunity in context. Immunity. 2004;21:315-29
9. Galban CJ, Galban S, Van Dort ME, Luker GD, Bhojani MS, Rehemtulla A. et al. Applications of molecular imaging. Prog Mol Biol Transl Sci. 2010;95:237-98
10. Kang JH, Chung JK. Molecular-genetic imaging based on reporter gene expression. J Nucl Med. 2008;49(Suppl 2):164S-79S
11. Ray P. The pivotal role of multimodality reporter sensors in drug discovery: from cell based assays to real time molecular imaging. Curr Pharm Biotechnol. 2011;12:539-46
12. Greer LF 3rd, Szalay AA. Imaging of light emission from the expression of luciferases in living cells and organisms: a review. Luminescence. 2002;17:43-74
13. Rabinovich BA, Ye Y, Etto T, Chen JQ, Levitsky HI, Overwijk WW. et al. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proc Natl Acad Sci U S A. 2008;105:14342-6
14. Loening AM, Fenn TD, Wu AM, Gambhir SS. Consensus guided mutagenesis of Renilla luciferase yields enhanced stability and light output. Protein Eng Des Sel. 2006;19:391-400
15. Chung E, Yamashita H, Au P, Tannous BA, Fukumura D, Jain RK. Secreted Gaussia luciferase as a biomarker for monitoring tumor progression and treatment response of systemic metastases. PLoS One. 2009;4:e8316
16. Santos EB, Yeh R, Lee J, Nikhamin Y, Punzalan B, La Perle K. et al. Sensitive in vivo imaging of T cells using a membrane-bound Gaussia princeps luciferase. Nat Med. 2009;15:338-44
17. Brust P, Haubner R, Friedrich A, Scheunemann M, Anton M, Koufaki ON. et al. Comparison of [18F]FHPG and [124/125I]FIAU for imaging herpes simplex virus type 1 thymidine kinase gene expression. Eur J Nucl Med. 2001;28:721-9
18. Campbell DO, Yaghoubi S, Su Y, Lee JT, Auerbach MS, Herschman H. et al. Structure-guided engineering of human thymidine kinase 2 as a positron emission tomography reporter gene for enhanced phosphorylation of a non-natural thymidine analog reporter probe. J Biol Chem. 2012
19. Ponomarev V, Doubrovin M, Shavrin A, Serganova I, Beresten T, Ageyeva L. et al. A human-derived reporter gene for noninvasive imaging in humans: mitochondrial thymidine kinase type 2. J Nucl Med. 2007;48:819-26
20. Kesarwala AH, Prior JL, Sun J, Harpstrite SE, Sharma V, Piwnica-Worms D. Second-generation triple reporter for bioluminescence, micro-positron emission tomography, and fluorescence imaging. Mol Imaging. 2006;5:465-74
21. Ponomarev V, Doubrovin M, Serganova I, Vider J, Shavrin A, Beresten T. et al. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging. 2004;31:740-51
22. Ray P, Tsien R, Gambhir SS. Construction and validation of improved triple fusion reporter gene vectors for molecular imaging of living subjects. Cancer Res. 2007;67:3085-93
23. Chinnasamy N, Shaffer J, Chinnasamy D. Production of multicistronic HIV-1 based lentiviral vectors. Methods Mol Biol. 2009;515:137-50
24. Azadniv M, Dugger K, Bowers WJ, Weaver C, Crispe IN. Imaging CD8+ T cell dynamics in vivo using a transgenic luciferase reporter. Int Immunol. 2007;19:1165-73
25. Chewning JH, Dugger KJ, Chaudhuri TR, Zinn KR, Weaver CT. Bioluminescence-based visualization of CD4 T cell dynamics using a T lineage-specific luciferase transgenic model. BMC Immunol. 2009;10:44
26. Byrne WL, Mills KH, Lederer JA, O'Sullivan GC. Targeting regulatory T cells in cancer. Cancer Res. 2011;71:6915-20
27. Cao X. Regulatory T cells and immune tolerance to tumors. Immunol Res. 2010;46:79-93
28. Jeremiah NM, Liston A. Genetic tools for analysis of FoxP3+ regulatory T cells in vivo. Methods Mol Biol. 2011;707:105-18
29. Hume DA. Applications of myeloid-specific promoters in transgenic mice support in vivo imaging and functional genomics but do not support the concept of distinct macrophage and dendritic cell lineages or roles in immunity. J Leukoc Biol. 2011;89:525-38
30. Creusot RJ, Yaghoubi SS, Chang P, Chia J, Contag CH, Gambhir SS. et al. Lymphoid-tissue-specific homing of bone-marrow-derived dendritic cells. Blood. 2009;113:6638-47
31. Tanaka M, Swijnenburg RJ, Gunawan F, Cao YA, Yang Y, Caffarelli AD. et al. In vivo visualization of cardiac allograft rejection and trafficking passenger leukocytes using bioluminescence imaging. Circulation. 2005;112:I105-10
32. Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunol Rev. 2011;239:27-44
33. Cao YA, Wagers AJ, Beilhack A, Dusich J, Bachmann MH, Negrin RS. et al. Shifting foci of hematopoiesis during reconstitution from single stem cells. Proc Natl Acad Sci U S A. 2004;101:221-6
34. Edinger M, Cao YA, Verneris MR, Bachmann MH, Contag CH, Negrin RS. Revealing lymphoma growth and the efficacy of immune cell therapies using in vivo bioluminescence imaging. Blood. 2003;101:640-8
35. Na IK, Markley JC, Tsai JJ, Yim NL, Beattie BJ, Klose AD. et al. Concurrent visualization of trafficking, expansion, and activation of T lymphocytes and T-cell precursors in vivo. Blood. 2010;116:e18-25
36. Schimmelpfennig CH, Schulz S, Arber C, Baker J, Tarner I, McBride J. et al. Ex vivo expanded dendritic cells home to T-cell zones of lymphoid organs and survive in vivo after allogeneic bone marrow transplantation. Am J Pathol. 2005;167:1321-31
37. Dubey P, Su H, Adonai N, Du S, Rosato A, Braun J. et al. Quantitative imaging of the T cell antitumor response by positron-emission tomography. Proc Natl Acad Sci U S A. 2003;100:1232-7
38. Koehne G, Doubrovin M, Doubrovina E, Zanzonico P, Gallardo HF, Ivanova A. et al. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nat Biotechnol. 2003;21:405-13
39. Marodon G, Mouly E, Blair EJ, Frisen C, Lemoine FM, Klatzmann D. Specific transgene expression in human and mouse CD4+ cells using lentiviral vectors with regulatory sequences from the CD4 gene. Blood. 2003;101:3416-23
40. Mouly E, Dorival C, Pflumio F, Baillou C, Coulombel L, Levy Y. et al. CD4 regulation in human lymphoid non-T-cells: a role for the silencer element. Mol Immunol. 2007;44:267-75
41. Pipkin ME, Ljutic B, Cruz-Guilloty F, Nouzova M, Rao A, Zuniga-Pflucker JC. et al. Chromosome transfer activates and delineates a locus control region for perforin. Immunity. 2007;26:29-41
42. Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E. et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255-6
43. Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P. et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415-9
44. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H. et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143-50
45. Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA. et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996;2:216-23
46. Rosenzweig M, Connole M, Glickman R, Yue SP, Noren B, DeMaria M. et al. Induction of cytotoxic T lymphocyte and antibody responses to enhanced green fluorescent protein following transplantation of transduced CD34(+) hematopoietic cells. Blood. 2001;97:1951-9
47. Stripecke R, Carmen Villacres M, Skelton D, Satake N, Halene S, Kohn D. Immune response to green fluorescent protein: implications for gene therapy. Gene Ther. 1999;6:1305-12
48. Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F. et al. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109:4708-15
49. Shu CJ, Guo S, Kim YJ, Shelly SM, Nijagal A, Ray P. et al. Visualization of a primary anti-tumor immune response by positron emission tomography. Proc Natl Acad Sci U S A. 2005;102:17412-7
50. Peggs KS, Allison JP. Co-stimulatory pathways in lymphocyte regulation: the immunoglobulin superfamily. Br J Haematol. 2005;130:809-24
51. Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611-4
52. Ponomarev V, Doubrovin M, Lyddane C, Beresten T, Balatoni J, Bornman W. et al. Imaging TCR-dependent NFAT-mediated T-cell activation with positron emission tomography in vivo. Neoplasia. 2001;3:480-8
53. Saibil SD, Deenick EK, Ohashi PS. The sound of silence: modulating anergy in T lymphocytes. Curr Opin Immunol. 2007;19:658-64
54. Patel MR, Chang YF, Chen IY, Bachmann MH, Yan X, Contag CH. et al. Longitudinal, noninvasive imaging of T-cell effector function and proliferation in living subjects. Cancer Res. 2011;70:10141-9
55. Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299-308
56. Dobrenkov K, Olszewska M, Likar Y, Shenker L, Gunset G, Cai S. et al. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. J Nucl Med. 2008;49:1162-70
57. Gade TP, Hassen W, Santos E, Gunset G, Saudemont A, Gong MC. et al. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080-8
58. Koya RC, Mok S, Comin-Anduix B, Chodon T, Radu CG, Nishimura MI. et al. Kinetic phases of distribution and tumor targeting by T cell receptor engineered lymphocytes inducing robust antitumor responses. Proc Natl Acad Sci U S A. 2010;107:14286-91
59. Roszik J, Rabinovich B, Cooper LJ. Imaging of T cells expressing chimeric antigen receptors. Immunotherapy. 2011;3:1411-4 doi:10.2217/imt.11.138
60. Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J. et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6:53-8
61. Radu CG, Shu CJ, Nair-Gill E, Shelly SM, Barrio JR, Satyamurthy N. et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2'-deoxycytidine analog. Nat Med. 2008;14:783-8
62. Namavari M, Chang YF, Kusler B, Yaghoubi S, Mitchell BS, Gambhir SS. Synthesis of 2'-deoxy-2'-[18F]fluoro-9-beta-D-arabinofuranosylguanine: a novel agent for imaging T-cell activation with PET. Mol Imaging Biol. 2011;13:812-8
Author contact
![]() Corresponding author: pdubeyedu
Corresponding author: pdubeyedu