Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Radionuclides and bifunctional...
Integrin αvβ3 targeted...
Radionuclide therapy targeting...
Conclusions
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2011; 1:201-210. doi:10.7150/thno/v01p0201 This volume Cite
Review
Integrin Targeted Delivery of Radiotherapeutics
Zhaofei Liu1,2 ![]() , Fan Wang1,2, Xiaoyuan Chen3
, Fan Wang1,2, Xiaoyuan Chen3 ![]()
1. Medical Isotopes Research Center, Peking University, Beijing, China
2. Department of Radiation Medicine, School of Basic Medical Sciences, Peking University, Beijing, China
3. National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institute of Health (NIH), Bethesda, MD
Published 2011-3-1
Abstract
Targeted radionuclide therapy, which is based on the selective delivery of a sufficient radiation dose to tumors without significantly affecting normal tissues, is a promising therapeutic approach for the treatment of a wide variety of malignancies. Integrins, a family of cell adhesion molecules, play key roles during tumor angiogenesis and metastasis. Among all the integrins, αvβ3 seems to be the most important in the process of tumor angiogenesis. Integrin αvβ3 is highly expressed on activated endothelial cells, new-born vessels as well as some tumor cells, but is not present in resting endothelial cells and most normal organ systems, making it a suitable target for anti-tumor therapy. In this review, we summarize the current development and applications of antibody-, peptide-, and other ligand-based integrin targeted radiotherapeutics for tumor radiation therapy.
Keywords: Cancer, integrin, radionuclide, radioimmunotherapy (RIT), peptide receptor radionuclide therapy (PRRT)
Introduction
Tumor angiogenesis, the sprouting of new blood vessels from preexisting vasculature, is well recognized as an essential mechanism for tumor growth and development of metastasis [1, 2]. Without the formation of neovasculature to provide oxygen and nutrients, tumors cannot grow beyond about 1~2 mm in size [3, 4]. Once vascularized, previously dormant tumors begin to grow rapidly, invade surrounding tissues (invasion), and transfer to distant sites in the body (metastasis). The angiogenic process depends on vascular endothelial cell migration and invasion, and is regulated by cell adhesion receptors. Integrins represent a subclass of cell adhesion molecules connecting the cytoskeleton with the extracellular matrix (ECM) or other cells. Integrins consist of two genetically nonrelated subunits, α and β, which are noncovalently associated with each other. In mammals, there are 18 α and 8 β subunits capable of assembling at least 24 different functional heterodimers [5-7]. Members of the integrin family play vital roles in the regulation of cellular activation, migration, proliferation, survival, and differentiation [8, 9]. Among all of the integrins, integrin αvβ3 has been identified as the most important member with overexpression pattern among vascular cells during tumor angiogenesis and vascular remodeling [1, 10, 11]. Integrin αvβ3 is highly expressed on activated endothelial cells and new-born vessels, but is absent in resting endothelial cells and most normal organ systems, making it a suitable target for anti-angiogenic cancer therapy. In addition, it is also expressed on some tumor cells, allowing for both tumor cell and tumor vasculature targeted therapy. To date, numerous anti-angiogenic therapies based on integrin αvβ3 antagonism, including antibodies, peptides, small molecules, small interfering RNA (siRNA) have been investigated [12].
Targeted delivery of radionuclides by tumor-specific ligands (antibodies, peptides, or small proteins) can specifically deliver radiation to tumors, while sparing the normal organs and tissues. The radiation energy given off by the radionuclides would also kill the adjacent tumor cells, which do not express the target antigen (so-called “crossfire”). In recent years, tumor targeted radionuclide therapy restimulates the interests of physicians especially after the successful clinical applications of the two Food and Drug Administration (FDA) approved antibodies (Zevalin and Bexxar) for radioimmunotherapy (RIT) of non-Hodgkin's lymphoma (NHL) [13]. Although RIT of solid tumors has shown less progress, a series of novel tumor targeted radiotherapeutic agents with favorable in vivo pharmacokinetics and enhanced tumor-to-nontumor ratios have been investigated in preclinical studies, and some of them are tested in clinical trials. In this article, we will first introduce the radionuclides and bifunctional chelators that are being used for tumor targeted radionuclide therapy, and then summarize the current development of integrin-targeted radiotherapeutics.
Radionuclides and bifunctional chelators
A tumor targeted radionuclide therapeutic agent is typically composed of the radionuclide and the targeting ligand (antibodies, peptides, or small proteins). For direct radio-iodination (with 131I, 125I or 123I), the iodine-ligand complex can be easily prepared. However, almost all metal radionuclides require chelation chemistry for attachment to the ligand. Bifunctional chelators (BFCs) that possess specific functional groups allow both conjugation to ligands and stable complex formation with metal radionuclides.
Therapeutic radionuclides
The suitability of a radionuclide for radiation therapy depends on its physical and chemical properties and the nature of the radiation, such as low or high linear energy transfer (LET) emission. The most commonly used radionuclides in tumor targeted therapy are β-emitters, although Auger electron-emitting radionuclides and α-emitters are also being used (Table 1) [14].
Selected radionuclides useful for tumor targeted radiotherapy
| Nuclide | Emission | Half-life | Emax (MeV) | Mean range (mm) | Source | Imageable |
|---|---|---|---|---|---|---|
| 90Y | β | 2.7 d | 2.30 | 2.76 | generator | No |
| 131I | β, γ | 8.0 d | 0.81 | 0.4 | reactor | Yes |
| 177Lu | β, γ | 6.7 d | 0.50 | 0.28 | reactor | Yes |
| 186Re | β, γ | 3.8 d | 1.1 | 0.92 | accelerator or reactor | Yes |
| 188Re | β, γ | 17.0 h | 2.1 | 2.43 | generator | Yes |
| 67Cu | β, γ | 2.6 d | 0.57 | 0.6 | accelerator | Yes |
| 213Bi | α | 45.7 min | 5.87 | 0.04-0.1 | generator | Yes |
| 212Bi | α | 1.0 h | 6.09 | 0.04-0.1 | generator | Yes |
| 211At | α | 7.2 h | 5.87 | 0.04-0.1 | accelerator | Yes |
| 67Ga | Auger, β, γ | 3.3 d | 0.18 | 0.001-0.02 | accelerator | Yes |
| 111In | Auger, γ | 2.83 d | 0.86 | 0.001-0.02 | accelerator | Yes |
131I and 90Y are the two most widely used radionuclides in clinical practice today. 131I is readily available, inexpensive, and can also provide γ-imaging emissions, which makes it possible for monitoring the therapeutic efficacy during the period of radiation therapy. However, the conventional conjugation of 131I to antibodies results in rapid degradation and a reduced residence time in the tumor, thus diminishing the tumor dose [15]. 90Y is a more energetic pure β-emitter and thus has fewer environmental radiation restrictions. 90Y possesses greater emission range and most of the decay energy is deposited in tumors only if their diameter is 1 cm or more [13], which makes 90Y more suitable for irradiation of larger tumors. Since 90Y is a pure β-emitter, 111In is usually chosen as the surrogate for imaging and dosimetry determination. 177Lu is an isotope with lower energy and longer half-life compared to 90Y. 177Lu has an imageable γ emission and this property also allows tracking the radiolabeled agents during therapy procedures by using external gamma scintigraphy. Rhenium isotopes (186Re and 188Re) have also been used for RIT, and have sufficient γ-energies for external scintigraphy, similar to 131I. 67Cu remains an interesting candidate for therapy with regards to emission energy, half-life and imageable emissions. Based on the good results of preclinical and clinical evaluations of 67Cu-labeled antibodies, broader clinical investigations in radioimmunotherapy trials are desirable. However, the availability of the 67Cu nuclide is a limiting factor for its more widespread use. Efforts to develop efficient procedures to produce large amounts of 67Cu with high specific activity would be much more helpful [16].
Radiation therapy with α-emitters has received renewed interest recently, especially with bismuth nuclides, such as 212Bi and 213Bi as eluates from 234Ra and 225Ac generators, respectively [17]. The cyclotron-produced radiohalogen 211At is also a promising candidate for RIT applications on the basis of half-life (t1/2 =7.2 h). The α-particle RIT is best used when there are micrometastases or circulating tumor cells, not bulky disease, because of their high LET and short effective path length in tissues [18]. Such high LET radiation has profound effects on DNA, causing strand breaks. Low-energy Auger electron-emitters are also used as alternative to α- or β-emitters for RIT. Most Auger electrons travel nanometer to micrometer distances in tissue and have high LET values approaching those of α-emitters (4-26 keV/μm) [19]. These properties render Auger electron-emitters highly cytotoxic and damaging to DNA when they decay intracellularly, especially when they decay in close proximity to the cell nucleus [20]. Studies have demonstrated that Auger emitters, such as 67Ga and 111In, might have a significant role as therapeutics, even if their clinical use might be limited to irradiation of microscopic residual disease [21].
Bifunctional Chelators
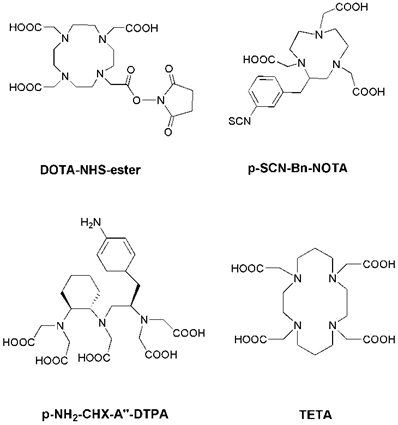
Radiolabeling with the radiometals is performed by means of chelation with chelators (Figure 1). Radioiodinated tyrosine is considered to be excreted from the cell after internalization, where the chelated radiometals (such as 90Y or 177Lu) metabolites are trapped in the lysosomes, thereby increasing the retention time of the isotope within the tumor [13]. DTPA (diethylene triamine pentaacetic acid) can chelate 111In, and 111In-DTPA-Octreotide (OctreoScan) is a commonly used agent in clinical application [22]. DTPA and derivatives usually lead to fast reaction kinetics [23]. DOTA (1,4,7,10-tetraazacyclododecane-1, 4, 7,10-tetraacetic acid) is a bifunctional chelator for the complexation of various diagnostic radioisotopes, such as 64Cu, 68Ga, 86Y and 111In, but also for the complexation of therapeutic radioisotopes, such as 67Cu, 177Lu and 90Y [24, 25]. DOTA is able to form stable complexes with divalent and trivalent metals. NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid) and TETA (1,4,8,11-tetraazacyclododenane-1,4,8,11-tetraacetic acid) are macrocyclic pyazapolycarboxylate chelators, which are characterized by a higher stability than DOTA for 64Cu labeling in vivo [26, 27].
Chemical structures of some common bifunctional chelators. DOTA = 1,4,7,10-tetraazacyclododecane-1, 4, 7,10-tetraacetic acid; NOTA = 1,4,7-triazacyclononane-1,4,7-triacetic acid; DTPA = diethylene triamine pentaacetic acid; TETA = 1,4,8,11-tetraazacyclododenane-1,4,8,11-tetraacetic acid.
Integrin αvβ3 targeted radionuclide therapy
The crucial roles of integrin αvβ3 in tumor angiogenesis have led to a promising strategy to block its signaling by antagonists, as this would theoretically inhibit the tumor angiogenesis or enhance the efficacy of other tumor therapeutics. In addition, the high expression of integrin αvβ3 on tumor new-blood vessels and some tumor cells makes the integrin αvβ3 a suitable maker for cancer-targeted drug delivery [5, 12]. Several delivery vehicles such as antibodies, RGD peptides, peptidomimetics, and other small molecules have been investigated for integrin targeted delivery of chemical drugs, cytotoxicities and gene inhibitors [12]. Integrin αvβ3 targeted radionuclide therapy of tumors by use of antibodies and RGD peptides was also investigated in the last decades.
Antibody-based radiotherapeutics targeting integrin αvβ3
The targeted systemic delivery of radiation to tumors through radiolabeled antibodies (radioimmunotherapy) offers several potential advantages over external beam radiotherapy, including the ability to specifically target multiple sites of disease, avoid or minimize normal tissue toxicity, and cause cell death of adjacent tumor cells. Preclinical and clinical investigations with murine mAbs highlighted several issues that require attention before successful applications in cancer management. Foremost of these issues was the inevitable production of human antimurine immunoglobulin antibodies (HAMA) after one to three treatments in patients. Some other factors limiting treatment include inadequate therapeutic dose delivered to tumor lesions, slow blood clearance, high uptake in normal organs, and insufficient tumor penetration. To date, this efforts such as the production of chimeric mAbs, grafting of complementarity-determining region (CDR) or complete humanization of the protein have primarily been applied to eliminate HAMA [28].
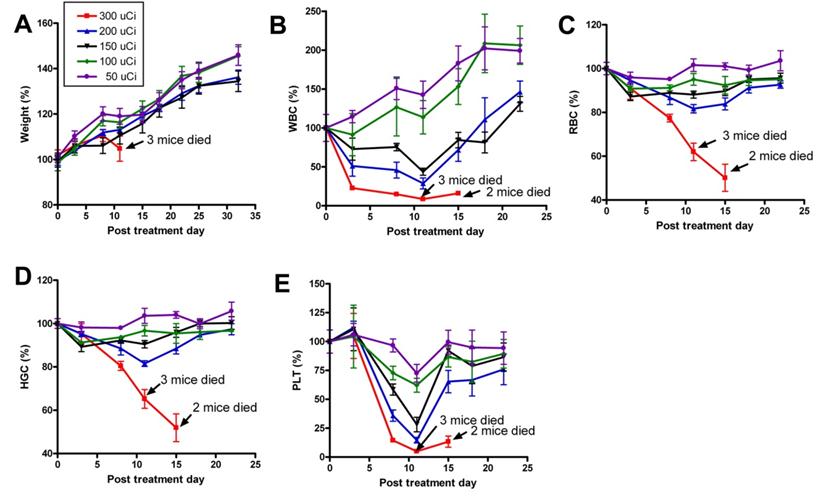
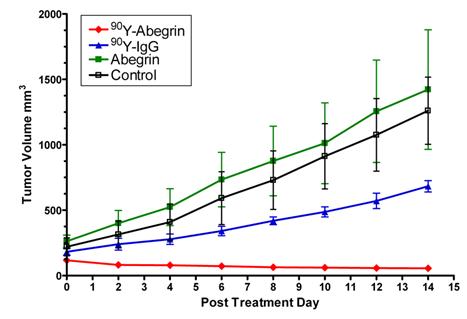
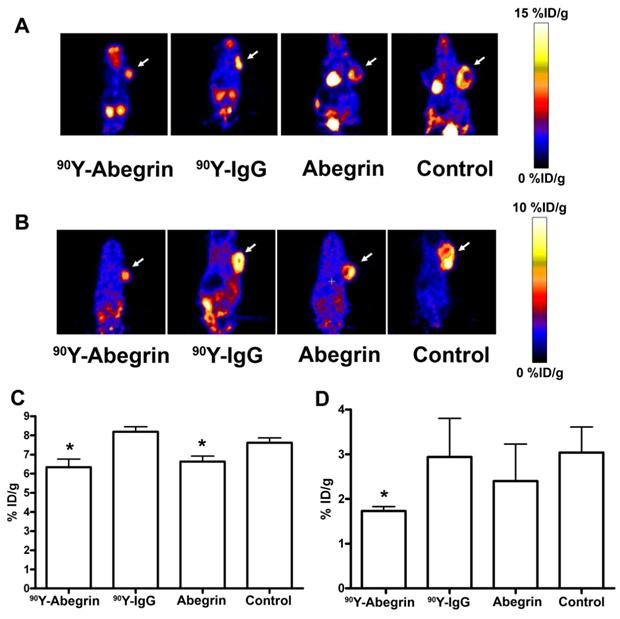
Recently, we prepared a 90Y-labeled humanized anti-integrin αvβ3 monoclonal antibody AbegrinTM and evaluated the RIT efficacy in U87MG glioblastoma xenograft models [29]. Maximum tolerated dose (MTD) and dose response analysis revealed 200 μCi per mouse as appropriate treatment dose with hepatic clearance and no organ toxicity (Figure 2). 90Y-Abegrin showed partial tumor regression with a final fractional tumor volume (Vfinal/Vinitial) of 0.69, as compared with that of 3.76 for 90Y-hIgG and 5.43 for normal AbegrinTM controls, respectively (Figure 3). [18F]-fluorodeoxyglucose (18F-FDG) microPET imaging revealed a reduction of cell proliferation and metabolic activity whereas 3'-[18F]fluoro-3'-deoxythymidine (18F-FLT) reflected decreased DNA synthesis in the 90Y-AbegrinTM group (Figure 4A-D). Ex vivo histological analysis also confirmed the therapeutic efficacy of 90Y-AbegrinTM. It was concluded that radioimmunotherapy with 90Y-labeled AbegrinTM may prove promising in the treatment of highly vascular, invasive, and heterogeneous malignant brain tumors [29].
A maximum tolerated dose (MTD) study was completed using escalating 90Y-AbegrinTM of 50,100,150, 200, and 300 μCi. Each dose was tested in seven female athymic nude mice. (A) Body weight changes of animals. (B-E) Animals that received 300 μCi suffered from hematologic toxicity with a decline in WBC (B), RBC (C), HGC (D), and platelet counts (E), and eventual mortality. Animals that received 50, 100, 150, or 200 μCi of activity did not experience significant reductions in WBC, RBC, HGC, or platelet counts. Adapted with permission from [29].
90Y-AbegrinTM dose response and inhibition of human glioblastoma growth in vivo. Nude mice bearing U87MG tumors were injected with a one-time dose of 100 μCi of 90Y-AbegrinTM, 90Y-IgG, AbegrinTM, or saline. The growth inhibition of experimental groups was monitored via serial caliper measurements. 90Y-Abegrin treatment animals maintained a statistically significant reduction in tumor size beginning on posttreatment day 2 and eventually showed partial tumor regression whereas all other groups showed increased final fractional tumor volumes. Adapted with permission from [29].
(A-B) Representative coronal microPET images and radioactivity accumulation quantification of female athymic nude mice bearing U87MG tumors (treated with 90Y-AbegrinTM, 90Y-IgG, AbegrinTM, or saline) after i.v. injection of 18F-FDG (A) and 18F-FLT (B). (C) 18F-FDG imaging revealed a statistically significant reduction in both 90Y-AbegrinTM and AbegrinTM signal intensity, suggesting reduced metabolic activity. (D) 18F-FLT imaging showed reduced tumor accumulation value in 90Y-AbegrinTM group, reflecting reduced DNA synthesis. Adapted with permission from [29].
The strategy to overcome the problems of intact antibodies, such as slow blood clearance, high background uptake, insufficient tumor penetration, has been the development of small molecular constructs, such as antibody fragments (e.g., Fab' and F(ab')2) and subfragments (e.g., scFv, (sFv)2), which are capable of binding to the tumor while clearing from normal tissues rapidly [28]. However, the tumor residence time, which is important for delivering therapeutic radiation doses, also significantly decreases as the immunoglobulin fragment becomes smaller. The pretargeting strategies that separate tumor targeting from delivery of the therapeutic radionuclide are also being considered to design optimized RIT agents. However, up to now, there are not extensive investigations of integrin targeted cancer radioimmunotherapy by use of such antibody fragments or pretargeting delivery systems.
RGD peptide-based radiotherapeutics targeting integrin αvβ3
Peptides are usually classified as containing less than 50 amino acids, ~5500 Da. This low molecular weight renders peptides low in antigenicity, fast in clearance, and rapid in tissue and tumor penetration. In contrast to monoclonal antibodies, automated techniques allow peptides to be produced easily and inexpensively [30]. In recent years, a wide variety of peptides have been identified with high affinity for characteristic receptors that are overexpressed on a large number of tumor cell types.
Since integrin αvβ3 binds a wide range of ECM molecules (such as fibronectin, fibrinogen, von Willebrand factor, vitronectin, and proteolysed forms of collagen and laminin) with an Arg-Gly-Asp (RGD) triplepeptide motif [29, 31], RGD peptides and analogues were therefore chemically synthesized to mimic the structure of the natural ligands of integrins and used as the integrin αvβ3 targeting vehicles. The RGD triple-peptide itself is limited in the in vivo use because of its short circulation half-life. Conformational restriction by ring closure of the peptides and further chemical modification, such as the use of D-amino acids, like in the c(RGDfV) (with f standing for D-phenylalanine) compound, not only increased their αvβ3 binding affinity, but also improved their bioavailability [32]. In the last decades, a series of radiolabeled cyclic RGD peptides and analogues have been intensively investigated for positron emission tomography (PET) and single photon emission computed tomography (SPECT) imaging of integrin αvβ3 expression [5, 33, 34]. However, only a small fraction of reports cover therapeutic tumor targeting. Janssen et al. [35] studied the in vivo behavior of the radiolabeled dimeric RGD peptide E[c(RGDfK)]2 in a subcutaneous (s.c.) ovarian carcinoma nude mouse model. The dimeric peptide E-[c(RGDfK)]2 labeled with 111In, 90Y and 99mTc, respectively. Tumor uptake was as high as 7.5 %ID/g (111In-DOTA-[c(RGDfK)]2) at 2 h p.i. or 6.0 %ID/g (99mTc-HYNIC-E-[c(RGDfK)]2) at 1 h p.i. A single injection of 37 MBq of 90Y-DOTA-E[c(RGDfK)]2 in mice with small s.c. tumors caused a significant growth delay as compared with control mice. Treatment with 37 MBq of 90Y-DOTA-E[c(RGDfK)]2 caused significant increased survivals as compared to mice treated with 37 MBq 90Y-labeled control peptide or untreated mice (median survival of 54 versus 33.5 versus 19 days, respectively). Unfortunately, in a follow-up study, increasing the number of injections did not improve the therapeutic efficacy [36]. Moreover, the prominent renal uptake of this conjugate limited its potential in clinical applications.
It has been proposed by others and us that the receptor binding characteristics of dimeric and multimeric RGD peptides would be better than that of monomeric RGD peptide based upon polyvalency [24, 33, 35, 37, 38]. The receptor binding of the one RGD peptide will significantly enhance the local concentration of the other RGD peptide in the vicinity of the receptor, which may lead to a faster rate of receptor binding or a slower rate dissociation of the dimeric RGD probes. The dimeric or multimer RGD peptide with almost one order of magnitude higher integrin binding affinity than the monomeric analog, and thus the dimeric or multimer RGD probes gave the highest tumor specific activity accumulation at all time points examined as compared to monomeric RGD peptide probes. Multimeric RGD peptides with even higher receptor affinity and longer tumor retention time might be more suitable for clinical translation. We therefore used 90Y-labeled tetrameric RGD peptides for integrin αvβ3-targeted internal radiotherapy of athymic nude mice tumor xenografts. 90Y-labeled tetrameric RGD were more effective in inhibiting integrin-positive tumor growth than 90Y-labeled dimeric RGD, due to the significantly increased tumor uptake [39]. However, the whole body toxicity of 90Y-RGD tetramer was also significantly higher than that of the same dose of 90Y-RGD dimer because 90Y-RGD tetramer also exhibited high uptake in normal organs especially the kidneys [39] .
We and our collaborators have recently developed a series of new RGD dimers with PEG4 and Gly3 linkers [40-45]. The insertion of the Gly3 or PEG4 spacers significantly increased the distance between the two cyclic RGD peptide motifs, resulting in an increase in vitro receptor-binding affinity. Importantly, the radiolabeled new types of RGD dimers (i.g. 3PRGD2) possessed as high tumor uptake as RGD tetramer (RGD4), but the uptake in normal organs was much lower compared with RGD tetramer due to the improved in vivo kinetics [39-41], which led to a lower toxicity and much higher maximum tolerated dose (MTD) of 90Y-DOTA-3PRGD2 in mice [39]. Significant anti-tumor vasculature effects can be found in the 90Y-DOTA-3PRGD2 treatment group. Compared to 90Y-DOTA-RGD4, the low accumulation of 90Y-DOTA-3PRGD2 in normal organs makes it more suitable for high dose or multiple-dose regimens, in order to achieve maximum therapeutic efficacy for integrin αvβ3-positive tumors [39].
In another report, considering that monomeric RGD peptides have a lower molecular mass compared with antibody or multimeric RGD peptides, Yoshimoto et al. [46] proposed that 90Y-labeled RGD monomer would be a promising radiopharmaceuticals for tumor therapy causing low radioactive exposure to normal tissues such as kidney and liver. The tumor therapeutic and imaging potential of 90Y/111In-labeled monomeric RGD peptide was investigated in a human ovarian carcinoma mouse model, and it was claimed that the RGD monomer can be used for fractionated therapy without evident toxicity. The radionuclide therapy results demonstrated that multiple dose administration of 90Y-DOTA-c(RGDfK) (3 × 11.1 MBq) led to an increased tumor growth inhibition in comparison to the single-dose administration (11.1 MBq). However, due to the lower tumor uptake of the RGD monomer, the single-dose administration did not show significant inhibition to tumor growth and the radionuclide therapeutic efficacy of the multiple dose administration was also generally limited, and the optimized regimens should be considered to reach the better results.
Others
Integrin targeted delivery of internal radiotherapy by non-peptide antagonists has also been reported. 90Y and 177Lu labeled TA138, a DOTA-conjugated non-peptide integrin αvβ3 antagonist, were prepared under anaerobic conditions to protect them from radiolytic degradation [47]. These complexes were synthesized in high yield and specific activity and showed high affinity for integrin αvβ3. 111In-TA138 was also synthesized for tumor imaging purposes as well as for dosimetry determination of 90Y-TA138 [48, 49]. High tumor uptake and low background activity of 111In-TA138 were found in the c-neu oncomouse mammary adenocarcinoma model (9.39 % ID/g at 2 h p.i.). Despite the differences in lipophilicity and solution structure between 90Y-TA138 and 111In-TA138, biodistribution studies showed that 111In-TA138 and 90Y-TA138 are biologically equivalent with respect to the uptakes in tumors and other major organs, indicating that 111In-TA138 was useful as an imaging surrogate for 90Y-TA138 and could predict the radiation dosimetry of 90Y-TA138. Radiotherapy using 90Y-TA138 in the c-neu oncomouse model demonstrated a slowing of tumor growth at a dose of 15 mCi/m2, and a regression of tumors at a dose of 90 mCi/m2 [48].
Knottin peptides are small constrained polypeptides that share a common disulfide-bonded framework and a triple-stranded β-sheet fold [50]. Knottin family members possess one or more surface-exposed loops that tolerate much sequence diversity, and different binding motifs could potentially be engineered into these loops to create bioactive knottins against different molecular targets [51, 52]. Several knottin mutants that bind to integrin receptors (αvβ3/αvβ5 or αvβ3/αvβ5/α5β1) with low nanomolar affinity have been identified [51, 52], and radionuclide and optical dye labeled such knottin peptides have demonstrated favorable in vivo tumor targeting properties [53-56]. Recently, two knottin peptides (2.5D and 2.5F: targeting integrin αvβ3/αvβ5 and αvβ3/αvβ5/α5β1, respectively) were radiolabeled with a therapeutic radionuclide 177Lu, and the resulting radiopharmaceuticals were evaluated for potential radiotherapy in a mouse model of human glioma [57]. Compared to 177Lu-DOTA-2.5D, 177Lu-DOTA-2.5F showed much higher tumor uptake and tumor to blood ratios, as well as a higher tumor to kidney radiation absorbed dose ratio, demonstrating the more promising application of 177Lu-DOTA-2.5F as a targeted radionuclide therapeutic agents for integrin-positive tumors.
Radionuclide therapy targeting other integrins
Although integrin αvβ3 has been extensively studied as one of the key players in tumor angiogenesis, other integrin members such as integrin α2β1, α3β1, α4β1, αvβ5 and αvβ6 are involved in these processes as well. Comparing with integrin αvβ3, the literature reports of other integrins targeted molecular imaging and drug delivery are relatively rare.
A high-affinity peptidomimetic ligand (LLP2A; IC50 = 2 pM) against α4β1 integrin was identified by using both diverse and highly focused one-bead-one-compound combinatorial peptidomimetic libraries in conjunction with high-stringency screening [58]. LLP2A was demonstrated that can be used to image α4β1-expressing lymphomas with high sensitivity and specificity when conjugated to a near infrared fluorescent dye in a mouse xenograft model. Thus, LLP2A shows great potential as an imaging and therapeutic agent for α4β1-positive tumors [58]. In the subsequent studies, the near infrared fluorescent dye Cy5.5, 64Cu and 111In labeled LLP2A was investigated for optical imaging, microPET and whole-body autoradiography (WBAR) of tumors, respectively [59, 60]. The s.c. tumors can be clearly visualized after i.v. injection of the conjugates, which warrants further investigation of the LLP2A conjugates as agents for α4β1 targeted imaging and therapy of human lymphoid malignancies. Unfortunately, to date, the integrin α4β1 targeted radionuclide therapy for tumors in preclinical and clinical investigations have not been reported. A peptide NAVPNLRGDLQVLAQKVART (denoted as A20FMDV2), derived from foot-and-mouth disease virus, has been identified as a potent inhibitor of αvβ6 [61], which is low or undetectable in most adult tissues but is up-regulated dramatically in many carcinoma tumors [62]. A20FMDV2 was radiolabeled with 18F and tested in mice bearing both αvβ6-negative and αvβ6-positive tumor xenografts [63]. Rapid uptake and selective retention of radioactivity in the αvβ6-positive tumor, together with the fast renal elimination of non-specifically bound activity, resulted in receptor specific imaging of the αvβ6-positive neoplasm with good contrast. To further improve the tumor targeting property and increase the retention in the target tissue, two PEGylated A20FMDV2 variants were prepared and both showed significantly improved retention in two αvβ6-expressing human tumor xenograft models, making them promising for molecular imaging of integrin αvβ6 expression [64]. However, for targeted radionuclide therapy of tumors, the PEGylated A20FMDV2 tracers may have limitations due to the low tumor uptake values (less than 3 %ID/g). To develop radiopharmaceuticals for α3β1 integrin targeting, an all D-amino acid analog of the residues 531-542 from the α1 chain of type IV collagen (which binds to α3β1 integrin) was synthesized by solid-phase methods, and then labeled with 64Cu [65]. The tumor accumulation of the tracer was very low (< 2 %ID/g) and blocking studies failed to reduce the tumor uptake, confirming that the low tumor uptake was mostly non-specific accumulation. The combination of the results obtained from the in vitro and in vivo data strongly suggest that peptides of this class targeted to the α3β1 would not be suitable as in vivo imaging agents in humans [65].
Radiotherapeutics targeting integrins besides αvβ3 have not been well investigated may largely due to the lack of high affinity/specificity ligands to each integrin. Therefore, ligand screening strategies, such as phage display, may play a major role in identifying novel ligands for each integrin. The optimizing strategies, such as multivalency [33] and PEGylation [66], may also be involved in the development of optimized ligands, which may open up new perspectives for cancer therapy based on integrin targeted radiotherapeutics.
Conclusions
Integrins are the key regulators of tumor angiogenesis and metastasis. The vast number of literature reports on anti-angiogenic cancer therapy based on integrin antagonism confirmed the validity of integrin αvβ3 as an anti-cancer target. However, the investigation of integrin αvβ3 targeted delivery of radiotherapeutics is relatively rare. Integrin targeted radionuclide therapy is considered to specifically deliver radiation to the tumor cells or tumor vasculature, thereafter leading to the death of tumor cells and the inhibition of tumor growth. Integrin αvβ3 targeted RIT with 90Y-labeled humanized antibody AbegrinTM was investigated in human glioblastoma xenografts and it was demonstrated the promising results for anti-tumor therapy. However, the radiation uptake in normal organs especially in the liver and spleen was high due to the slow circulation clearance and liver excretion of the intact antibody. RGD peptides that specifically targeting integrin αvβ3 were also investigated for radionuclide therapy of tumors with rapid blood clearance and optimized tumor penetration. However, the tumor inhibition efficacies of RGD peptides-based radiotherapeutics were nonoptimized due to the lower tumor uptake. Therefore, further research effort is still needed to develop novel integrin targeted radiotherapeutics with better tumor targeting efficacy and desirable pharmacokinetics. In addition, the combination of integrin targeted radiation therapy with other therapeutic modalities, such as chemotherapy, is also expected to generate significantly greater anti-tumor benefits.
Acknowledgements
Research carried out in the authors' laboratories was supported in part by the Intramural Research Program (IRP) of the National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institutes of Health (NIH) and National Natural Science Foundation of China grants 81000625, 81028009, 30870728, 30930030, and 30900373; an “863” project grant 2007AA02Z467; a "973" project 2011CB707703; Ministry of Science and Technology of China grants 2009ZX09103-733, 2009ZX09301-010 and 2009ZX09103-746.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401-10
2. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249
3. Sharma RA, Harris AL, Dalgleish AG, Steward WP, O'Byrne KJ. Angiogenesis as a biomarker and target in cancer chemoprevention. Lancet Oncol. 2001;2:726-32
4. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27-31
5. Cai W, Niu G, Chen X. Imaging of integrins as biomarkers for tumor angiogenesis. Curr Pharm Des. 2008;14:2943-73
6. Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673-87
7. Cai W, Wu Y, Chen K, Cao Q, Tice DA, Chen X. In vitro and in vivo characterization of 64Cu-labeled Abegrin, a humanized monoclonal antibody against integrin αvβ3. Cancer Res. 2006;66:9673-81
8. Cooper CR, Chay CH, Pienta KJ. The role of αvβ3 in prostate cancer progression. Neoplasia. 2002;4:191-4
9. Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91-100
10. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin αvβ3 for angiogenesis. Science. 1994;264:569-71
11. Eliceiri BP, Cheresh DA. The role of αv integrins during angiogenesis: insights into potential mechanisms of action and clinical development. J Clin Invest. 1999;103:1227-30
12. Liu Z, Wang F, Chen X. Integrin αvβ3-targeted cancer therapy. Drug Develop Res. 2008;69:329-39
13. Milenic DE, Brady ED, Brechbiel MW. Antibody-targeted radiation cancer therapy. Nat Rev Drug Discov. 2004;3:488-99
14. Dixon KL. The radiation biology of radioimmunotherapy. Nucl Med Commun. 2003;24:951-7
15. Goldenberg DM. Targeted therapy of cancer with radiolabeled antibodies. J Nucl Med. 2002;43:693-713
16. Novak-Hofer I, Schubiger PA. Copper-67 as a therapeutic nuclide for radioimmunotherapy. Eur J Nucl Med Mol Imaging. 2002;29:821-30
17. McDevitt MR, Sgouros G, Finn RD, Humm JL, Jurcic JG, Larson SM. et al. Radioimmunotherapy with alpha-emitting nuclides. Eur J Nucl Med. 1998;25:1341-51
18. McDevitt MR, Ma D, Lai LT, Simon J, Borchardt P, Frank RK. et al. Tumor therapy with targeted atomic nanogenerators. Science. 2001;294:1537-40
19. Buchegger F, Perillo-Adamer F, Dupertuis YM, Delaloye AB. Auger radiation targeted into DNA: a therapy perspective. Eur J Nucl Med Mol Imaging. 2006;33:1352-63
20. Boswell CA, Brechbiel MW. Auger electrons: lethal, low energy, and coming soon to a tumor cell nucleus near you. J Nucl Med. 2005;46:1946-7
21. Delbaldo C, Raymond E, Vera K, Hammershaimb L, Kaucic K, Lozahic S. et al. Phase I and pharmacokinetic study of etaracizumab (Abegrin), a humanized monoclonal antibody against αvβ3 integrin receptor, in patients with advanced solid tumors. Invest New Drugs. 2008;26:35-43
22. van der Lely AJ, de Herder WW, Krenning EP, Kwekkeboom DJ. Octreoscan radioreceptor imaging. Endocrine. 2003;20:307-11
23. Shi J, Liu Z, Jia B, Yu Z, Zhao H, Wang F. Potential therapeutic radiotracers: preparation, biodistribution and metabolic characteristics of 177Lu-labeled cyclic RGDfK dimer. Amino Acids. 2010;39:111-20
24. Iten F, Muller B, Schindler C, Rochlitz C, Oertli D, Macke HR. et al. Response to [90Yttrium-DOTA]-TOC treatment is associated with long-term survival benefit in metastasized medullary thyroid cancer: a phase II clinical trial. Clin Cancer Res. 2007;13:6696-702
25. Mohsin H, Fitzsimmons J, Shelton T, Hoffman TJ, Cutler CS, Lewis MR. et al. Preparation and biological evaluation of 111In-, 177Lu- and 90Y-labeled DOTA analogues conjugated to B72.3. Nucl Med Biol. 2007;34:493-502
26. Prasanphanich AF, Nanda PK, Rold TL, Ma L, Lewis MR, Garrison JC. et al. [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues. Proc Natl Acad Sci U S A. 2007;104:12462-7
27. Liu Z, Li ZB, Cao Q, Liu S, Wang F, Chen X. Small-animal PET of tumors with 64Cu-labeled RGD-bombesin heterodimer. J Nucl Med. 2009;50:1168-77
28. Milenic DE. Radioimmunotherapy: designer molecules to potentiate effective therapy. Semin Radiat Oncol. 2000;10:139-55
29. Veeravagu A, Liu Z, Niu G, Chen K, Jia B, Cai W. et al. Integrin αvβ3-targeted radioimmunotherapy of glioblastoma multiforme. Clin Cancer Res. 2008;14:7330-9
30. Weiner RE, Thakur ML. Radiolabeled peptides in the diagnosis and therapy of oncological diseases. Appl Radiat Isot. 2002;57:749-63
31. Van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285-98
32. Eble JA, Haier J. Integrins in cancer treatment. Curr Cancer Drug Targets. 2006;6:89-105
33. Liu S. Radiolabeled multimeric cyclic RGD peptides as integrin αvβ3 targeted radiotracers for tumor imaging. Mol Pharm. 2006;3:472-87
34. Chen X. Multimodality imaging of tumor integrin αvβ3 expression. Mini Rev Med Chem. 2006;6:227-34
35. Janssen M, Oyen WJ, Massuger LF, Frielink C, Dijkgraaf I, Edwards DS. et al. Comparison of a monomeric and dimeric radiolabeled RGD-peptide for tumor targeting. Cancer Biother Radiopharm. 2002;17:641-6
36. Janssen M, Frielink C, Dijkgraaf I, Oyen W, Edwards DS, Liu S. et al. Improved tumor targeting of radiolabeled RGD peptides using rapid dose fractionation. Cancer Biother Radiopharm. 2004;19:399-404
37. Dijkgraaf I, Kruijtzer JA, Liu S, Soede AC, Oyen WJ, Corstens FH. et al. Improved targeting of the αvβ3 integrin by multimerisation of RGD peptides. Eur J Nucl Med Mol Imaging. 2007;34:267-73
38. Wu Y, Zhang X, Xiong Z, Cheng Z, Fisher DR, Liu S. et al. microPET imaging of glioma integrin αvβ3 expression using 64Cu-labeled tetrameric RGD peptide. J Nucl Med. 2005;46:1707-18
39. Liu Z, Shi J, Jia B, Yu Z, Liu Y, Zhao H. et al. 90Y-labeled two multimeric RGD peptides RGD4 and 3PRGD2 for integrin targeted radionuclide therapy. Mol Pharm. [Epub ahead of print]
40. Liu Z, Liu S, Wang F, Liu S, Chen X. Noninvasive imaging of tumor integrin expression using 18F-labeled RGD dimer peptide with PEG4 linkers. Eur J Nucl Med Mol Imaging. 2009;36:1296-307
41. Shi J, Wang L, Kim YS, Zhai S, Liu Z, Chen X. et al. Improving tumor uptake and excretion kinetics of 99mTc-labeled cyclic arginine-glycine-aspartic (RGD) dimers with triglycine linkers. J Med Chem. 2008;51:7980-90
42. Shi J, Kim YS, Zhai S, Liu Z, Chen X, Liu S. Improving tumor uptake and pharmacokinetics of 64Cu-labeled cyclic RGD peptide dimers with Gly3 and PEG4 linkers. Bioconjug Chem. 2009;20:750-9
43. Wang L, Shi J, Kim YS, Zhai S, Jia B, Zhao H. et al. Improving tumor-targeting capability and pharmacokinetics of 99mTc-labeled cyclic RGD dimers with PEG4 linkers. Mol Pharm. 2009;6:231-45
44. Liu Z, Liu S, Niu G, Wang F, Liu S, Chen X. Optical imaging of integrin αvβ3 expression with near-infrared fluorescent RGD dimer with tetra(ethylene glycol) linkers. Mol Imaging. 2010;9:21-29
45. Liu S. Radiolabeled cyclic RGD peptides as integrin αvβ3-targeted radiotracers: maximizing binding affinity via bivalency. Bioconjug Chem. 2009;20:2199-213
46. Yoshimoto M, Ogawa K, Washiyama K, Shikano N, Mori H, Amano R. et al. αvβ3 Integrin-targeting radionuclide therapy and imaging with monomeric RGD peptide. Int J Cancer. 2008;123:709-15
47. Liu S, Harris TD, Ellars CE, Edwards DS. Anaerobic 90Y- and 177Lu-labeling of a DOTA-conjugated nonpeptide vitronectin receptor antagonist. Bioconjug Chem. 2003;14:1030-7
48. Harris TD, Kalogeropoulos S, Nguyen T, Liu S, Bartis J, Ellars C. et al. Design, synthesis, and evaluation of radiolabeled integrin αvβ3 receptor antagonists for tumor imaging and radiotherapy. Cancer Biother Radiopharm. 2003;18:627-41
49. Onthank DC, Liu S, Silva PJ, Barrett JA, Harris TD, Robinson SP. et al. 90Y and 111In complexes of a DOTA-conjugated integrin αvβ3 receptor antagonist: different but biologically equivalent. Bioconjug Chem. 2004;15:235-41
50. Pallaghy PK, Nielsen KJ, Craik DJ, Norton RS. A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994;3:1833-9
51. Kimura RH, Levin AM, Cochran FV, Cochran JR. Engineered cystine knot peptides that bind αvβ3, αvβ5, and α5β1 integrins with low-nanomolar affinity. Proteins. 2009;77:359-69
52. Kimura RH, Cheng Z, Gambhir SS, Cochran JR. Engineered knottin peptides: a new class of agents for imaging integrin expression in living subjects. Cancer Res. 2009;69:2435-42
53. Kimura RH, Miao Z, Cheng Z, Gambhir SS, Cochran JR. A dual-labeled knottin peptide for PET and near-infrared fluorescence imaging of integrin expression in living subjects. Bioconjug Chem. 2010;21:436-44
54. Jiang L, Kimura RH, Miao Z, Silverman AP, Ren G, Liu H. et al. Evaluation of a 64Cu-labeled cystine-knot peptide based on agouti-related protein for PET of tumors expressing αvβ3 integrin. J Nucl Med. 2010;51:251-8
55. Miao Z, Ren G, Liu H, Kimura RH, Jiang L, Cochran JR. et al. An engineered knottin peptide labeled with 18F for PET imaging of integrin expression. Bioconjug Chem. 2009;20:2342-7
56. Nielsen CH, Kimura RH, Withofs N, Tran PT, Miao Z, Cochran JR. et al. PET imaging of tumor neovascularization in a transgenic mouse model with a novel 64Cu-DOTA-knottin peptide. Cancer Res. 2010;70:9022-30
57. Jiang L, Miao Z, Kimura RH, Liu H, Cochran JR, Culter CS. et al. Preliminary evaluation of 177Lu-labeled knottin peptides for integrin receptor-targeted radionuclide therapy. Eur J Nucl Med Mol Imaging. 2010 [Epub ahead of print]
58. Peng L, Liu R, Marik J, Wang X, Takada Y, Lam KS. Combinatorial chemistry identifies high-affinity peptidomimetics against α4β1 integrin for in vivo tumor imaging. Nat Chem Biol. 2006;2:381-9
59. Peng L, Liu R, Andrei M, Xiao W, Lam KS. In vivo optical imaging of human lymphoma xenograft using a library-derived peptidomimetic against α4β1 integrin. Mol Cancer Ther. 2008;7:432-7
60. Denardo SJ, Liu R, Albrecht H, Natarajan A, Sutcliffe JL, Anderson C. et al. 111In-LLP2A-DOTA Polyethylene glycol-targeting α4β1 integrin: comparative pharmacokinetics for imaging and therapy of lymphoid malignancies. J Nucl Med. 2009;50:625-34
61. DiCara D, Rapisarda C, Sutcliffe JL, Violette SM, Weinreb PH, Hart IR. et al. Structure-function analysis of Arg-Gly-Asp helix motifs in αvβ6 integrin ligands. J Biol Chem. 2007;282:9657-65
62. Bates RC. The αvβ6 integrin as a novel molecular target for colorectal cancer. Future Oncol. 2005;1:821-8
63. Hausner SH, DiCara D, Marik J, Marshall JF, Sutcliffe JL. Use of a peptide derived from foot-and-mouth disease virus for the noninvasive imaging of human cancer: generation and evaluation of 4-[18F]fluorobenzoyl A20FMDV2 for in vivo imaging of integrin αvβ6 expression with positron emission tomography. Cancer Res. 2007;67:7833-40
64. Hausner SH, Abbey CK, Bold RJ, Gagnon MK, Marik J, Marshall JF. et al. Targeted in vivo imaging of integrin αvβ6 with an improved radiotracer and its relevance in a pancreatic tumor model. Cancer Res. 2009;69:5843-50
65. Edwards WB, Anderson CJ, Fields GB, Welch MJ. Evaluation of radiolabeled type IV collagen fragments as potential tumor imaging agents. Bioconjug Chem. 2001;12:1057-65
66. Walsh S, Shah A, Mond J. Improved pharmacokinetics and reduced antibody reactivity of lysostaphin conjugated to polyethylene glycol. Antimicrob Agents Chemother. 2003;47:554-8
Author contact
![]() Corresponding author: Dr. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institute of Health (NIH), 31 Center Dr, 31/1C22, Bethesda, MD 20892, USA; Tel: 301-451-4246; Email: shawn.chengov; Dr. Zhaofei Liu, Medical Isotopes Research Center, Peking University, 38 Xueyuan Road, Beijing 100191, China; Tel: 86-10-82802871; Email: liuzfedu.cn
Corresponding author: Dr. Xiaoyuan Chen, Laboratory of Molecular Imaging and Nanomedicine (LOMIN), National Institute of Biomedical Imaging and Bioengineering (NIBIB), National Institute of Health (NIH), 31 Center Dr, 31/1C22, Bethesda, MD 20892, USA; Tel: 301-451-4246; Email: shawn.chengov; Dr. Zhaofei Liu, Medical Isotopes Research Center, Peking University, 38 Xueyuan Road, Beijing 100191, China; Tel: 86-10-82802871; Email: liuzfedu.cn